2.1.1 Définition
Les descripteurs moléculaires servent à extraire
et définir un ensemble d'informations concernant les
caractéristiques des molécules. Ils sont liés
implicitement ou explicitement aux
propriétés physico-chimiques de ces
dernières. Les informations liées avec
ces
caractéristiques seront traduites ensuite en une série de
grandeurs (en général scalaires) (Dudek, 2006). Ces grandeurs
sont appelés des descripteurs et sont déterminés pour
chaque molécule et ensuite liées mathématiquement à
l'activité biologique mesurée (Figure 2) (Kier,
1975). Un descripteur peut être parfois plus complexe comme par exemple
en cas de champs d'interaction dans l'espace 3D. Dans ce dernier cas, les
molécules sont superposées et alignées sur une grille et
les potentiels d'interaction sont déterminés pour chaque
molécule au niveau de chaque point de grille (CONSONNI, 2002).
En d'autres termes, les descripteurs sont nombreux et de
différentes complexités et de conceptions diverses.
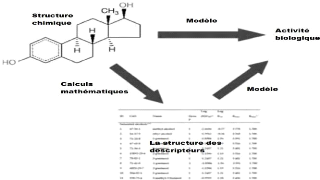
Figure 2. Des descripteurs moléculaires relient
la structure chimique à l'activité
biologique.
Le modèle QSAR/QSPR est une fonction
mathématique qui a comme paramètres les descripteurs
moléculaires et un objectif principal qui consiste à
déterminer la fonction biologique d'un produit chimique à partir
de sa structure.
5
2.1.2 Types de descripteurs
Dans la littérature, de nombreux descripteurs
moléculaires ont été introduits, plus de 10 000
descripteurs moléculaires qui quantifient des caractéristiques
physico-chimiques ou structurelles de molécules (Karelson, 2002). En
générale, ces descripteurs peuvent être obtenus par deux
façons différentes : empirique ou non-empirique, mais les
descripteurs calculés et non mesurés sont les plus
préférés, car ils répondent bien aux objectifs de
la modélisation puisque ils permettent des prédictions sans
passer par une étape de synthèse. Au contraire, il y a quelques
descripteurs mesurés qui utilisent généralement des
données expérimentales (polarisabilité, ou potentiel
d`ionisation...). La classification des descripteurs se fait fréquemment
en fonction de la dimensionnalité de la représentation
moléculaire sur laquelle ils sont calculés: on parlera donc de
descripteurs 0D, 1D, 2D et 3D. Malgré leur origine et leur
représentation mathématique différentes, la plupart des
descripteurs moléculaires sont interconnectés. Par exemple, les
changements topologiques des molécules peuvent être liés
aux changements de leur géométrie. Cependant, les changements au
niveau de la symétrie ou de la ramification peuvent affecter la
distribution de la charge électronique, ce qui peut causer des
changements dans la réactivité chimique ou la polarité,
respectivement. De même, dans certains cas, un paramètre
décrivant la distribution de la densité électronique au
sein de la molécule pourrait agir comme une meilleure mesure de
ramification, et donc de symétrie et de forme, ce qui est
irréalisable par les descripteurs topologiques typiques (Bauer,
1988).
a. Les descripteurs 0D
Tous les descripteurs moléculaires de cette classe 0D
ne nécessitent aucune information sur la structure moléculaire.
Les nombres d'atomes et de liaisons, ainsi que la somme ou la moyenne des
propriétés atomiques sont typiques de cette classe de
descripteurs. Ces descripteurs sont calculés, interprétés
facilement et ne nécessitent pas d'optimisation de la structure
moléculaire. Ils montrent généralement une très
forte dégénérescence, c'est-à-dire qu'ils ont des
valeurs égales pour plusieurs molécules, telles que les
isomères. Malgré que leur contenu informationnel est faible, mais
ils peuvent néanmoins jouer un rôle important dans la
modélisation de plusieurs propriétés physico-chimiques ou
prendre part à des modèles plus complexes.
b. Les descripteurs 1D
Cette catégorie de descripteurs est calculée
à partir de la formule brute de la molécule en utilisant la
composition moléculaire. Celle-ci nous permet de faire des calculs en
fonction des propriétés atomique de la molécule telles que
: les pourcentages massiques des atomes, la masse molaire, le poids
moléculaire etc. Il est noté MW et mesuré en daltons (Da).
Le poids molécule est définit sous forme de la somme des poids
atomiques des différents atomes constituant la molécule. Il est
utilisé dans l'étude de transport dont la diffusion et le mode de
fonctionnement. Plus les composés sont munis de poids
moléculaire, plus ils sont moins susceptibles d'être
absorbés. En effet, le fait de garder des poids moléculaires plus
bas que possible devrait être l'objectif pour établir un
médicament (Rekkab, 2014). Pour les médicaments
délivrés par voie orale le poids moléculaire doit
être inférieur ou égal à 500 daltons (optimum autour
de 300 daltons) (C.A. Lipinski, 1997).
6
Le pourcentage massique est défini par la formule
suivante : %massique =
Ces descripteurs montrent généralement une
dégénérescence moyenne plus élevée,
cependant ils sont très utiles dans la modélisation à la
fois des propriétés physico-chimiques et biologiques.
c. Les descripteurs 2D
Les descripteurs moléculaires 2D utilisent une
représentation sous forme de graphes dits «descripteurs 2D»
(ou indices topologiques). Dans cette catégorie on trouve principalement
les descripteurs topologiques qui contiennent des informations relatives
à la connectivité ainsi que des estimations des
propriétés physicochimiques. Ce sont des descripteurs plus riches
d'information qui permettent la prédiction de la majorité des
propriétés moléculaires. Les informations sur les
propriétés moléculaires peuvent inclure Le nombre de
liaisons, les mesures de branchement et les représentations
théoriques des graphes de la structure moléculaire sont des
descripteurs courants de ce type. Les descripteurs 2D sont
considérés un peu plus complexes en comparaison avec les
descripteurs 0D ou 1D. Ces descripteurs 2D sont généralement
calculés en ensembles et sont représentés sous forme de
tableaux (Figure 3) de nombres binaires, entiers ou
réels (Euler, 1976) et (Schultz, 1989). En se basant sur ces tableaux,
nous pouvons donc calculer de nombreux indices topologiques. Parmi ceux, que
nous pouvons calculer, on trouve par exemple:
? L`indice de Wiener (Wiener, 1947), cet
indice permet de calculer le nombre total de liaisons dans les chemins les plus
courts entre toutes les paires d`atomes (en excluant les hydrogènes). Il
est défini par la formule suivante :
?
Avec : N est le nombre des atomes de la molécule et est
le plus petit nombre de liaisons séparant les deux atomes i et j.
? L'indice de Balaban (Balaban, 1982),
noté (J), est l`un des indices topologiques les plus importants et qui
permet de décrire le degré de ramification des molécules
non cycliques. Il est défini par la formule suivante :
?( )
Avec : i et j sont les atomes voisins (autres que
l`hydrogène), N est le nombre d`atomes
de la molécule , et sont les connectivités des
atomes i et j, est le nombre des liaisons,
et est le nombre des cycles.
? La somme des degrés de valence (D.
Jaiswal, 2006) , notée (SVD), est définit sous forme de la somme
de tous les degrés de valence de la molécule
représentée par un graphe, le degré d`un point
correspondant au nombre de lignes se terminant par ce point. Ce
paramètre dépend donc principalement de la ramification de la
molécule.

7
Figure 3. Exemple de tableaux de
connectivité C et de distance D de la molécule de l`acridine
d. Les descripteurs 3D
Les descripteurs moléculaires 3D sont basés sur
l'utilisation des positions relatives des atomes dans l'espace. Ils peuvent
décrire des caractéristiques plus complexes ; leurs calculs
nécessitent donc de connaître la géométrie 3D de la
molécule en passant le plus souvent par une modélisation
moléculaire empirique ou ab-initio. Ces descripteurs présentent
un temps de calcul relativement coûteux, mais apportent davantage en
termes d'informations fournis. Ils sont nécessaires à la
modélisation de propriétés ou d'activités qui
dépendent de la structure 3D. On distingue plusieurs familles
importantes dans cette catégorie de descripteurs.
* Les descripteurs géométriques:
les plus populaires sont le volume moléculaire, la surface
accessible au solvant et le moment principal d'inertie.
* Les descripteurs électroniques:
permettent de quantifier différents types d'interactions inter-
et intramoléculaires liées à l'activité biologique
des molécules. Le calcul de la plupart
de ces descripteurs nécessite la recherche de la
géométrie pour laquelle l'énergie stérique est
minimale, et cela se fait souvent à l'aide de la chimie quantique. Par
exemple, les énergies de la plus haute orbitale moléculaire
occupée (HOMO) et de la plus basse vacante (LUMO) (orbitales
frontières) sont des descripteurs fréquemment
sélectionnés. Le moment dipolaire, le potentiel d'ionisation et
différentes énergies relatives à la molécule sont
d'autres paramètres importants.
* Les descripteurs spectroscopiques: en
utilisant ces descripteurs, les molécules peuvent être
caractérisées par des mesures spectroscopiques en fonction de
leurs ondes vibrationnelles. En effet, les vibrations d'une molécule
dépendent de la masse des atomes et des forces d'interaction entre
ceux-ci; ces vibrations fournissent donc des informations sur la structure de
la molécule et sur sa conformation. Les spectres infrarouges peuvent
être obtenus soit de manière expérimentale, soit par un
calcul théorique, après recherche de la géométrie
optimale de la molécule
e. Les descripteurs locaux des
propriétés surfaciques moléculaires
Ce descripteur a été introduit par les
chercheurs en se basant sur le principe d'échafaudage moléculaire
« Scaffold ». Ce principe est l'un des concepts les plus importants
et les plus largement utilisés en chimie médicinale. Il utilise
les propriétés moléculaires locales pour extraire des
informations à partir de leur nature surfacique et structurelle de
constitution (Figure 4). L'échafaudage
représente généralement les structures centrales du cadre
moléculaire. Dans la littérature, plusieurs définitions
ont été introduites à propos des échafaudages
moléculaires, mais la définition la plus largement adoptée
est donnée par (Bemis, et al., 1996), en obtenant l'échafaudage
par la suppression de toutes les chaînes latérales (ou groupes R).
Parmi tous les échafaudages introduits dans littérature, dans ce
chapitre, on s'intéresse plus spécifiquement à ceux ayant
des propriétés de bioactivité
préférées (qui sont appelés "échafaudages
privilégiés" (Barreiro, London, 2015) ou "échafaudages
bioactifs" (Varin, et al., 2011) et (Nakagawa, et al., 2018)) qui bien
sûr présentent un intérêt particulier pour la
découverte de médicaments. De plus, plusieurs études ont
démontré que ces échafaudages sont pertinents pour la
chimie computationnelle et médicinale. Par exemple, l'analyse
informatique vise à isoler et comparer systématiquement les
structures centrales des composés actifs.
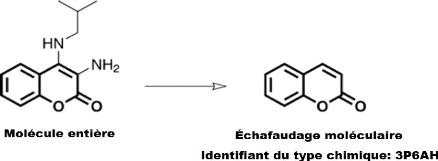
8
Figure 4. Exemple d'illustration d'échafaudage
moléculaire
f. Les descripteurs
quantiques/électroniques
Ces descripteurs s'intéressent à des
caractéristiques supplémentaires de la structure
moléculaire, surtout les informations moléculaires liées
avec la densité électronique résultante de distribution de
charge des molécules et ceux liées avec la chimie quantique sous
forme de données structurales, énergétiques,
électroniques et spectroscopiques (Mezey, 1993) et (Mezey, 1999) .
L'analyse de la densité électronique nous permet donc de
quantifier les différents types d'interactions inter et
intramoléculaires qui ont une relation potentille avec l'activité
biologique au niveau de la molécule.
| 

