|
|
|
Ministère de l'Enseignement Supérieur et
de la Recherche Scientifique
Université de Gafsa
Faculté des sciences de Gafsa
Unité de Recherche Matériaux,
Environnement et Energie (UR 14 ES 26)
|
|
|
MEMOIRE
Présenté par
AbdelKader MAJOURI
En vue de l'obtention du diplôme de
Mastère de recherche
Matériaux Innovants et Gestion
d'Energie
Intitulé
Etude ab initio des propriétés
électroniques et optiques des
couches minces à base de
Ti????
Soutenu le 00/00/2019, devant le jury composé de :
Mr Sghayer Taoufik Président
Mr Elarbi Tarek Rapporteur
Mr Khaldi Othmen Maître assistant à la FSG
Encadreur
Année Universitaire 2018 / 2019
|
|
Merci à tous
Remerciements
C
e travail a été réalisé au
département de physique de la Faculté des Sciences de Gafsa FSGF
et au Laboratoire des Matériaux-Organisation et Propriétés
(LMOP) à la Faculté des Sciences de Tunis-Campus Universitaire,
1060 Belvédère, Tunis El Manar- Tunisie.
En premier lieu, Je remercie ALLAH le
tout-puissant de m'avoir donné la confiance, la volonté et la
patience de mener à terme ce présent travail.
Je tiens tout d'abord à remercier vivement mon
encadreur Monsieur OTHMEN KHALDI pour m'avoir confié ce
travail, avoir dirigé l'ensemble de mon travail, pour son aide, ses
conseils avisés, et son expérience et intelligence qui m'a
été très précieux pour surmonter les
difficultés et améliorer la qualité du travail.
J'adresse toute ma gratitude aussi à Mr
TAREK ELARBI et tous les membres de laboratoire LMOP à la
faculté des sciences de Tunis pour l'aide et les conseils et
à Mr ********* et à Mr *********
qui ont accepté d'être rapporteurs de ce travail
malgré leurs nombreuses charges.
Enfin, Je remercie ma famille et mes chers amis pour leur
compréhension et soutien moral, j'espère que ce travail soit la
bonne expression.
AbdelKader
Dédicaces
Je dédie ce travail tout d'abord à
:
A ma chère mère, A mon cher
père,
Pour tous leurs sacrifices, leur amour, leur tendresse et
leur soutien durant l'élaboration de mon travail de fin
d'étude.
A mes frères,
Elakrmi, Amor, Tarek, Mohamed, Mohamed Elkhames et ma
belle-soeur Saida.
Je ne peux trouver les mots sincères pour vous
exprimer mon affection et mes pensées, vous étés pour moi
des frères sur qui je peux les compter et je vous souhaite un meilleur
avenir.
A mes amis,
Je tiens à remercier mes amies et tous ceux qui, de
près ou de loin, ont contribué à la réalisation de
ce travail.
Conclusion 18
Tables des matières
Tables des matières Liste
des figures Liste des tableaux
Introduction générale 1
Chapitre I : Généralités sur le
Ti02 et la Théorie de la Fonctionnelle de la Densité DFT
I. Généralités sur le dioxyde
de titane
1.1 Introduction 4
1.2 La phase rutile 4
1.3 La phase anatase 5
1.4 La phase brookite 6
1.5 Domaines d'applications de Ti02 7
II. Théorie de la Fonctionnelle de la
Densité DFT
2.1 Introduction 8
2.2 Densité électronique p (r) 9
2.3 Equation de Schrödinger 9
2.4 Approximation adiabatique de Born-Oppenheimer 10
2.5 Approximation de Hartree-Fock 11
2.6 Théorèmes de Hohenberg et Kohn 11
2.6.1 Premier théorème 12
2.6.2 Deuxième théorème 12
2.7 Les équations de Kohn et Sham 13
2.8 Fonctionnelle d'échange-corrélation
14
2.8.1 Approximation de la densité locale LDA
14
2.8.2 Approximation de gradient
généralisée GGA 15
2.8.3 Approximation de gradient
généralisée GGA-PBE et GGA+U 15
2.9 Fonctionnelle hydride B3LYP 16
2.10 Résolution des équations de Kohn et Sham
17
2.11 Cycle auto-cohérant de la résolution des
équations de Kohn et Sham 18
Chapitre II : Méthodes de calcul et
matériels
I. Technique d'élaboration des couches de
TiO2 : Spray pyrolyse
1.1 Le Spray pyrolyse 20
1.2 Système expérimental de Spray pyrolyse
20
1.2.1 Le spray nozzle 21
1.2.2 Pompe 21
1.2.3 Plaque chauffante
...................................................................................21
II. Logiciel Materials Studio
2.1 Présentation 22
2.2 Code CASTEP 24
2.2.1 Présentation 24
2.2.2 L'optimisation de la géométrie avec
CASTEP 25
2.2.3 Théorème de Bloch 25
2.2.4 Echantillonnage de la zone de Brillouin 26
2.2.5 Energie de coupure et super cellules 26
III. Dynamique Moléculaire
DM
3.1 Introduction 27
3.2 Les équations du mouvement 28
3.3 Algorithmes d'intégration numérique
29
3.3.1 Algorithme de Verlet 29
3.3.2 La symétrie par renversement du temps de Verlet
30
3.3.3 Algorithme de saute-mouton 30
3.4 Conditions périodiques CP 31
3.5 Boite de simulation 31
3.6 Potentiels 32
3.7 Calcul d'énergie et des forces 33
3.8 Thermostat 33
3.9 Déroulement de la simulation DM 33
3.9.1 Les conditions initiales 34
3.9.2 Equilibration 34
3.9.3 Production 35
Conclusion 35
Chapitre III : Résultats et discussions
3.1 Détail de calcul 37
3.2 Optimisation de la géométrie 37
3.3 Spectre Raman 41
3.4 Propriétés électroniques 44
3.4.1 Structure des bandes et densité d'états
électronique DOS 44
3.5 Propriétés optiques 49
3.5.1 Fonction diélectrique 50
3.5.2 Coefficient d'absorption 51
3.5.3 Indice de réfraction 54
3.6 Résultats du la dynamique moléculaire
56
3.6.1 Détail du calcul 56
3.6.2 Paramétres de mailles 59
3.6.3 Structure des bandes et densité d'état
électronique DOS 60
Conclusion générale
64
Bibliographie 66
Listes des figures
FIGURE 1.1. Le dioxyde de titane
sous forme de poudre 4
FIGURE 1.2. Structure de la phase
rutile de Ti02 5
FIGURE 1.3. Structure de la phase
anatase de Ti02 6
FIGURE 1.4. Structure de la phase
brookite de Ti02 6
FIGURE 1.5. Cycle
auto-cohérant de la résolution des équations de Kohn et
Sham 19
FIGURE 2.1. Schéma
général d'un procédé de dépôt par
pyrolyse par pulvérisation 22
FIGURE 2.2. Logiciel Materials
Studio 23
FIGURE 2.3. (A) Station de calcul,
(B) Logiciel Team Viewer 23
FIGURE 2.4. Code CASTEP dans
Materials Studio 24
FIGURE 2.5. Optimisation de la
géométrie avec CASTEP 25
FIGURE 2.6. Super cellule de la
phase anatase : (A) Super cellule (2 X 2 X 1)
et (B) Super
cellule (3 X 3 X 1)
26
FIGURE 2.7. Principe de
l'algorithme du Verlet-Leap-Frog 31
FIGURE 2.8. Représentation
schématique de la duplication de la boite de simulation à deux
dimensions. En utilisant les conditions aux limites périodiques,
lorsqu'une particule se déplace et quitte la boite principale, ses
images dans les cellules voisines se déplacent de la même
façon. 32
FIGURE 3.1. Structure cristalline
du dioxyde de titane anatase 37
FIGURE 3.2. Variation de la
convergence en fonction de l'énergie de coupure 38
FIGURE 3.3. (A) Structure avant
l'optimisation, (B) et (C) : Optimisation de la géométrie
du
TiO2 39
FIGURE 3.4. La convergence
d'optimisation avec CASTEP 40
FIGURE 3.5. L'optimisation de la
géométrie du TiO2 anatase en fonction d'énergie 40
FIGURE 3.6. Schéma des
déplacements des atomes pour les différentes modes des
vibrations
41
FIGURE 3.7. Spectres Raman
expérimentale du TiO2 anatase 42
FIGURE 3.8. Spectres Raman du TiO2
anatase DFT/B3LYP 44
FIGURE 3.9. Les structures des
bandes du TiO2 anatase 46
FIGURE 3.10. Les densités
d'état électronique DOS du TiO2 anatase 48
FIGURE 3.11. Fonction
diélectrique du TiO2 anatase 50
FIGURE 3.12. L'absorbance du
TiO2 anatase calculé par GGA-PBE 51
FIGURE 3.13. Le spectre
d'absorption et de réflexion expérimentale du TiO2 anatase
52
FIGURE 3.14.
Détermination de gap optique du TiO2 anatase
53
FIGURE 3.15. L'indice de
réfraction en fonction de l'énergie du TiO2 anatase 54
FIGURE 3.16. La structure
anatase en cours de calcul dynamique 57
FIGURE 3.17. Evolution de la
constante du mouvement en fonction du temps de simulation
57
FIGURE 3.18. Evolution de la
pression en fonction du temps de simulation 58
FIGURE 3.19. Evolution de la
température en fonction du temps de simulation 58
FIGURE 3.20. Statut de calcul
des paramétres de mailles 59
FIGURE 3.21. Les structures des
bandes pour deux pressions 61
FIGURE 3.22. Les densités
d'états électroniques DOS 62
Liste des tableaux
TABLEAU 1.1. Différences
entre les trois structures de Ti??2 7
TABLEAU 2.1. Ensembles
statistiques 34
TABLEAU 3.1. Variation de la
convergence en fonction des différentes valeurs d'énergie
de coupure 38
TABLEAU 3.2. (a)
Paramétres de maille de l'anatase, (b) Coordonnées
cartésiens 41
TABLEAU 3.3. Fréquences
des différentes bandes Raman de la phase anatase du TiO2 43
TABLEAU 3.4. Les valeurs de
l'énergie de gap obtenue [40] 49
TABLEAU 3.5. Fonction
diélectrique du TiO2 51
TABLEAU 3.6. Les
paramétres des mailles calculés et expérimentales
60
Introduction Générale
1 | P a g e
Introduction générale
L
e développement très rapide des technologies
liées à la microélectronique a accru la puissance des
ordinateurs permettant la mise en place de nouvelles techniques de
résolution numérique des problèmes physiques. Cette
puissance informatique de calcul a été encore optimisé,
des superordinateurs ont atteint le seuil de la «
suprématie quantique », ils sont capables de
reproduire les résultats de 50 qubits pendant 90 microsecondes.
Depuis longtemps, la simulation numérique est devenue
un outil classique pour d'écrire et caractériser les
évolutions et le comportement d'un systéme physique.
Aujourd'hui il y a plusieurs méthodes de simulation qui
représentent un outil de base pour le calcul des différentes
propriétés des systèmes les plus complexes, parmi ces
méthodes la méthode de calcul ab-initio. Ce calcul ab-initio est
très puissant et basé sur la théorie de la fonctionnelle
de la densité (DFT).
Cette mémoire est dédiée à
l'étude des propriétés des couches minces à base de
(TiO2). L'importance technologique de ces couches impose la connaissance de
leurs paramétres de bandes tels que les propriétés
structurales, électroniques et optiques.
La simulation est effectuée au sein de la
faculté des sciences de Gafsa par le code CASTEP (Cambridge Serial Total
Energy Package). Ce code est inclut dans le logiciel Materials Studio Modeling
basé sur la théorie de la fonctionnelle de la densité
(DFT).
Le but de ce travail est de calculer les
propriétés de couches minces à base de (TiO2)
et de corréler entre les résultats de simulations
théoriques calculées et expérimentales
observées.
Un autre objectif est réalisé au sein de la
faculté des sciences de Tunis au laboratoire LMOP, l'élaboration
des échantillons de TiO2 par la technique de spray pyrolyse dans le but
de déterminer les propriétés otiques de ce matériau
(absorbance, réflectance...).
Cette étude rentre dans le cadre de l'utilisation des
capacités MIN dans les mémoires ReRAM dans le domaine de la
microélectronique.
Dans le premier chapitre, nous avons
détaillé quelques généralités sur le dioxyde
de titane ainsi leurs domaines d'applications et nous avons
énoncé les principes de la théorie de la fonctionnelle de
la densité (DFT).
Dans le deuxième chapitre, nous avons
présenté la méthode d'élaboration des couches
minces et tous les matériels et les équipements
utilisés.
2 | Page
Introduction générale
Le troisième chapitre résume
nos résultats, leurs interprétations ainsi qu'une comparaison
avec des travaux expérimentales.
Nous terminerons ce travail par une synthèse globale
des résultats obtenus, donnée sous forme de conclusion
générale.
« Peu importe combien votre théorie est
belle, peu importe votre
intelligence, peu importe si vous
étés célèbre... Si votre théorie n'est pas
en
accord avec l'expérience, elle est fausse. C'est tout.
»
Richard Feynman, grand théoricien de la
physique quantique
Chapitre I
Généralités sur le Ti???? et la
théorie
de la fonctionnelle de la densité DFT
Chapitre 1 Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
4 | Page
I. Généralités sur le dioxyde de
titane :
1.1 Introduction :
Le dioxyde de titane ou aussi oxyde de titane (IV) TiO2 a
été découvert sous sa forme ilménite sous le nom de
Menachite en 1791 par William Gregor dans la région des Cornouailles en
Grande Bretagne [1]. C'est un composé chimique blanc très
brillant (figure 1.1), chimiquement stable, non toxique et
moins couteux. Il se cristallise sous trois phases, phase rutile, phase anatase
et la phase brookite. Par contre, à d'autres conditions de
température et de pression nous pouvons le trouver sous 8 autres formes.
L'indice de réfraction est assez beaucoup élevé, [2] et
pratiquement insensible à la lumière visible à cause de sa
large bande interdite Eg = 3.2 eV que ne lui permet l'absorption que
dans le domaine ultraviolet UV. Cette propriété est beaucoup
utilisée pour fabriquer des produits de protection (les crèmes de
bronzage....) contre le rayonnement ultraviolet élevé émis
par le soleil, qui peut provoquer un risque pour la santé (coups de
soleil, caractères et cancers de la peau). Depuis longtemps, le TiO2 a
gagné un grand prestige notable dans les sciences des matériaux.
Cela est dû à ses propriétés physico-chimiques
importantes, ce qui a signé ses propres empreintes parmi les autres
matériaux dans la recherche scientifique.

FIGURE 1.1 - Le dioxyde de titane
sous forme de poudre
1.2 La phase rutile :
La phase rutile est la phase la plus dense et la forme la plus
stable à haute température et à haute pression [3].
Chapitre 1 Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
Cette structure possède une symétrie
tétragonal décrite par le groupe d'espace P42/mnm. Les
paramètres de maille sont les suivants : a = b = 4.587A et c = 2.954
.
Chaque atome de titane de cette structure se trouve au centre
d'un octaèdre formé d'atomes d'oxygène
légèrement distordu avec quatre liaisons équatoriales Ti-O
courtes (1,945 A) et deux liaisons apicales plus longues (1,979 A [4].
Le rutile est un isolant de bande interdite d'énergie
Eg = 3.02 eV, lorsqu'il est stoechiométrique. Par contre,
pour d'autres conditions il peut se présenter comme un semiconducteur de
type N [5]. La structure de la phase rutile est représentée sur
la figure 1.2.

5 | Page
FIGURE 1.2 - Structure de la
phase rutile de Ti??2
1.3 La phase anatase :
La phase anatase est la phase moins stable «
métastable » qui se forme à une température moins
basse que le rutile et la brookite. Cette phase est plus complexe que la phase
rutile. La maille élémentaire est de symétrie
tétragonal, et décrit par le groupe d'espace I41/amd.
Les
paramétres de maille sont respectivement les suivants :
a = b = 3.780 A et c = 9.510 A . Pour chaque atome de titane on a quatre
liaisons quasi-équatoriales courtes (1.933 A) et deux liaisons apicales
longues (1.978 A). L'anatase est un semi-conducteur de bande interdite 3.2 eV.
Un changement de phase peut se produire à une température de 820
°C [6]. La structure de la phase anatase est représentée sur
la figure (1.3).
Chapitre 1 Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
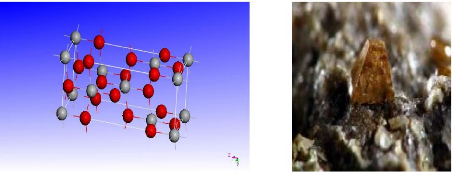
FIGURE 1.3 - Structure de la
phase anatase de Ti??2
1.4 La phase brookite :
Cette phase diffère aux autres phases rutile et anatase
par sa métastabilité et qu'il est difficile de l'obtenir en
laboratoire. Mais il est possible de la trouver comme une phase secondaire avec
l'anatase et le rutile [7].
La phase brookite possède une structure orthorhombique
qui appartient au groupe d'espace Pbca. Les paramètres de mailles sont
respectivement les suivants : a = 5.4558 A , b = 9.1819 A, et c = 5.1429 A .
Cette phase se forme à des températures basses que celle de la
phase rutile.

6 | Page
FIGURE 1.4 - Structure de la phase
brookite de Ti??2
Chapitre 1 Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
7 | Page
|
Phase
|
Phase rutile
|
Phase anatase
|
Phase brookite
|
|
Paramètres de
maille
(A°)
|
a=b=4.587 c=2.954
|
a=b=3.782 c=9.502
|
a=5.4558 b=9.1819, c=5.1429
|
|
La densité
(g.????-??)
|
4.20 à 5.60
|
3.82 à 3.97
|
4.17
|
|
Température de
fusion
(°C)
|
1830 à 1850
|
1843
|
1825
|
|
La masse
moléculaire
|
79.9
|
79.9
|
79.9
|
|
L'indice de
réfraction
|
2.75
|
2.54
|
2.586
|
|
La couleur
|
Brun, jaune, rouge, gris
|
Brun, bleu, noir, incolore
|
Brun foncée ou noir verdâtre
|
TABLEAU 1.1 - Différences
entre les trois structures de Ti??2
1.5 Domaines d'applications de Ti???? :
Aujourd'hui le dioxyde de titane est un matériau
semi-conducteur très intéressant. Il est utilisé dans
plusieurs domaines d'applications. On va citer ci-dessous quelques
applications.
y' Dans la technologie photocatalytique, le
TiO2 est essentiellement utilisé comme un photocatalyseur. Il
permet l'absorption des photons par les matériaux semi-conducteurs dans
les cellules photovoltaïques.
y' Dans la protection solaire, on sait que le
soleil émet un rayonnement ultra-violet, cette lumière noire
invisible peut engendrer un risque pour la santé (coups de soleil,
caractères et cancers de la peau). Cette rayonnement est
contrôlé par l'indice UV, plus il est élevé plus le
risque est fort. Alors grâce à l'indice de réfraction
important de TiO2, il peut absorber et bloquer les rayons UV de
spectre de soleil qui arrivent à la peau. Récemment des
crèmes solaires à base de TiO2 ont étés
utilisés pour la protection, c'est le bronzage.
Chapitre 1 Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
8 | Page
y' Dans le but de réduire la
pollution, le TiO2 nettoie l'air que nous respirons. Lorsque les
combustibles fossiles sont brulés par les véhicules, les
centrales électriques, les usines...des gaz sont libérés
dans l'atmosphère avec des conséquences très
néfastes sur l'air. Alors le TiO2 est utilisé pour purifier l'air
que nous respirons.
y' Dans les peintures et les
revêtements, le TiO2 est utilisé aussi en tant qu'un
pigment blanchisseur, il est appelé blanc de titane ou le plus blanc des
blancs, pigment blanc 5 ou CI 77891. Grace à ces qualités
blanchissantes pures et puissantes, il est utilisé comme un colorant
pour blanchir certains éléments comme les médicaments, les
bonbons, chewing-gum ...
y' Dans l'industrie alimentaire, le TiO2 est
utilisé comme un pigment blanchisseur très brillant. Sous forme
des nanoparticules il est utilisé en tant qu'additif alimentaire sous le
code E171 [8] dans plusieurs aliments comme les bonbons, chocolats,
gâteaux,... Ces additifs alimentaires sont des substances ajoutés
pour améliorer l'innocuité, la fraîcheur, le goût, la
texture ou l'aspect. Le TiO2 sous forme des nanoparticules est autorisé
dans les cosmétiques et dans les aliments, sauf en France où il
sera interdit dans l'industrie alimentaire dès le 1er janvier 2020. La
cause que le dioxyde de titane sous forme des nanoparticules l'E171 peut
être toxique et cancérigène. Cet additif est présent
dans le deux tiers des dentifrices et plusieurs autres éléments
et n'est pas indiqué sur l'emballage.
II. Théorie de la Fonctionnelle de la
Densité DFT :
2.1 Introduction :
Dernièrement il est trop difficile de décrire la
morphologie et la cristallographie des nanoparticules (les électrons,
les molécules...) par les lois de la mécanique classique.
La théorie de la fonctionnelle de la densité
« DFT » (en anglais Density Functionnal Theory) est
développée dans les années 1960 [9], est devenue un outil
pour la description et l'étude des propriétés physiques et
chimiques pour les systèmes contenant un grand nombre d'électrons
en utilisant les lois de la mécanique quantique.
Chapitre 1 Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
9 | Page
Les avances considérables dans les dix dernières
années, à la fois dans le domaine de la théorie, des
élaborations de cette théorie et la puissance informatique, ne
font que la modélisation de composés réels (à
structures complexes) devient parfaitement facile avec des moyens et un temps
de calcul rapide. Récemment elle est devenue parmi les méthodes
ab-initio les plus puissantes pour l'étude des systèmes de taille
importante.
On se basant sur les lois fondamentales de la mécanique
quantique, on peut déterminer les propriétés
électroniques et optiques d'un tel matériau à
l'état fondamental sans utiliser des variables extérieurs
ajustables. Ce calcul est appelé calcul ab-initio ou parfois
appelé calcul de premiers principes.
2.2 Densité électronique ?? (??) :
La densité électronique est la quantité
basique de la théorie et d'un point de vue personnel je désire de
la nommer « la clé de la théorie ».
Elle désigne tout simplement la probabilité de trouver l'un parmi
les N électrons dans l'élément de volume dr? et
s'exprime en n(r) par l'équation suivante :
Equation 1.1 ??( ?? ???) = N?
... . . . ? ?? |(??????? ... ... ... ...
????????) |?? d????
d???? ?????? ...
d????
Avec ñ( r ??) obeit a ces deux conditions
:
??( ????? ?8)=?? Et ? ??(?? ???)??????? = ??
2.3 Equation de Schrödinger :
La DFT permet d'exprimer toutes les propriétés
d'un système quantique en fonction de la densité
électronique ñ(r). Cette équation est indépendante
de temps, d'où la DFT est le cadre de la théorie quantique non
relativiste. Elle cherche à réduire un problème à N
corps (multicorps) à un problème à un seul corps
(monocorps) (ou biocorps si l'on considéré le spin). Ce passage
s'oppose à remplacer la fonction d'onde multiélectronique par la
densité électronique adéquate. La connaissance des
propriétés d'un système est reliée à la
connaissance exacte de son énergie totale, qui va être
calculée en résolvant l'équation de Schrödinger [10]
avec toutes les interactions entre les électrons et les ions.
Chapitre 1 Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
10 | Page
L'équation de Schrödinger est la suivante :
Equation 1.2 H??=
E??
Ou : H est l'opérateur hamiltonienne, ø sa fonction
d'onde et E l'énergie totale de systéme.
La solution de l'équation de Schrödinger conduit
à la résolution d'un problème à N corps, nous avons
besoin donc de faire des approximations pour faciliter cette
résolution.
H est l'opérateur Hamiltonien du système,
s'écrit comme suit :
Equation 1.3 H = ???? +
????-??(r) + ????-??(r, R)
+ ????+
????-??(R)
Avec :
Te : Terme d'énergie cinétique des
électrons. Ve-e : Terme d'interaction électrons-électrons.
Ve-n : Terme d'interaction électrons-noyaux. Tn : Terme
d'énergie cinétique des noyaux. Vn-n : Terme d'interaction
noyaux-noyaux.
2.4 Approximation adiabatique de Born-Oppenheimer :
Cette méthode a été publiée pour la
première fois en 1927 par Born et Oppenheimer BO [11]. Cette
approximation est très importante car elle permet alors de traiter
séparément la partie électronique et la partie
nucléaire.
A cause de la différence énorme entre la masse des
électrons et la masse des noyaux atomiques, elle consiste à
supposer que chaque électron se déplace indépendamment
dans un champ moyen crée par les autres électrons et noyaux
Donc il sera facile de négliger le mouvement des noyaux
à celles d'électrons et le problème à N corps sera
automatiquement réduit.
De ce fait les variables de l'équation de Schrödinger
seront uniquement les positions électroniques. Alors l'hamiltonien
totale peut alors être remplacé par l'hamiltonien
électronique suivante :
Chapitre I Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
11 | Page
Equation 1.4 ?????????????? = ???? + ????-?? +
????-??
Automatiquement l'équation de Schrödinger sera alors
aussi la suivante :
Equation 1.5 ??é????é??(??, R) =
??????????é??(??, R)
Maintenant le problème est purement électronique
ce qui donne à cette approximation le nom adiabatique (adiabatique :
dans un système les conditions externes permettent l'adaptation de
système, ce qui résulte en un changement de la densité de
probabilité).
2 .5 Approximation de Hartree-Fock :
Pour résoudre l'équation (1.5), les
méthodes de Hartree-Fock s'appliquent à des systèmes des
électrons homogènes basés sur l'hypothèse
d'électrons libres. Ces méthodes sont beaucoup utilisées
en chimie quantique ce qui est n'est pas le cas pour les atomes, solides et
molécules qui sont pratiquement inhomogènes.
2.6 Théorèmes de Hohenberg et Kohn :
Une fois la densité électronique est
définie, il est donc nécessaire de manipuler les lois de la DFT.
La théorie de la fonctionnelle de la densité est basée sur
deux théorèmes, ces théorèmes ont été
élaborés par Hohenberg en 1964 et Kohn en 1965 [12].
2.6.1 Premier théorème :
Un système à Ne
électrons en interaction et soumis à un
potentiel extérieur Vext. L'hamiltonien de
systéme s'écrit alors :
Equation 1.6 ??é?? = ?? + ?????? + ? ????
?????? (????)?????
?????
Chapitre I Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
12 | Page
Les travaux de Hohenberg et Kohn ont montrés que
l'énergie totale d'un gaz des électrons en présence d'un
potentiel extérieur est une fonctionnelle unique de la densité
électronique. Ce dernier est seul qui peut donner les
propriétés électroniques de systéme.
Equation 1.7 E = E[p(??)]
2.6.2 Deuxième théorème :
Il s'agit d'un théorème variationnel sur la
densité électronique [13]. La valeur minimale de cette
fonctionnelle est exactement la valeur de l'état fondamentale et que la
densité qui conduit à cette énergie est la densité
exacte de l'état fondamentale.
Equation 1.8 E(po) = ??????E(p)
ña : La
densité de l'état fondamental.
De ce fait l'énergie totale du système qui est une
fonctionnelle de la densité électronique prend la forme suivante
[14] :
Equation 1.9 E = E[p(??)] = ??[p(??)] + f ?????? (??)
p(??)????
F[ñ(r)] Est une fonction
universelle de ?? qui contient la contribution
cinétique et colombienne à l'énergie qui ne dépend
pas du système.
Le terme f Vex (r) ñ(r)dr
représente l'interaction entre noyau-électron. Une
fois si la fonctionnelle F[ñ(r)] est connue,
il faut utiliser le principe variationnel pour calculer l'énergie totale
et la densité électronique de l'état fondamentale.
Malheureusement, le théorème de Hohenberg et Kohn n'explicitent
pas la forme de F[ñ(r)].
Equation 1.10 ?????? [p] = To [p] + ??????
[p]
Chapitre I Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
13 | Page
2.7 Les équations de Kohn et Sham :
Le problème de N corps n'est pas encore résolu
à cause de l'interaction entre électron-électron. Pour
assouplir à ce problème Kohn et Sham ont proposé en 1965,
de substituer le systéme réel de particules en interaction par un
système fictif [15] sans interaction et qui possède la même
densité électronique que le système d'électrons en
interaction. Ils ont écrit la densité électronique comme
étant la somme des densités des particules libres :
Equation 1.11 ?????? [??] = ?? ?? [??] + ?????? [??] +
?????? [??]
Avec Equation 1.12 ??????[??] = ????
???(??)??'(??) |??-??'| ???? ????'
Equation 1.13 ???? [??] = ? ??????
?? ??=?? ?????|- ???? ?? |?????
L'expression de l'énergie devient alors maintenant :
Equation 1.14 ??[??] = ????[??] + ?????? [??] + ? ??????
(??) ??(??)???? + ??????[??]
Exc[ñ] : Energie
d'échange-corrélation qui décrit l'interaction
interélectronique.
Cette énergie est associée à un potentiel
d'échange-corrélation :
????????[??]
Equation 1.15 ?????? =
????
On doit résoudre maintenant facilement les
équations de Kohn et Sham.
-????
???? +????
???????? ? ??(??)
Equation 1.16 (
|??-??'| ???? + ?????? + ??????)???? =
????????
????
Equation 1.17 (-????
???? ???? + ????????)???? =
????????
Chapitre I Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
14 | Page
Avec ???????? = ????
???????? ? ??(??)
|??-??'|???? + ?? ???? + ?????? Equation 1.18
2.8 Fonctionnelle d'échange-corrélation :
L'interaction d'échange est un effet quantique
lié au principe de Pauli d'indiscernabilité des électrons
; l'énergie d'un système est calculée en tenant compte la
possibilité de permutation de d'indépendance entre particules.
La connaissance exacte de potentiel
d'échange-corrélation [16] signifie que nous avons résolue
exactement le problème de multicorps. La seule imprécision dans
l'approche de Kohn et Sham (KS) est le terme
d'échange-corrélation qui est inconnu. Cette fonctionnelle [17]
est une somme de deux parties, elle est très complexe ce que rend la
résolution des équations KS difficile. Néanmoins elle peut
être soumise à des approximations de l'ordre local ou proche local
de la densité.
2.8.1 Approximation de la densité locale LDA
:
L'approximation de la densité locale (en anglais Local
Density Approximation) est une méthode basée sur le modèle
de gaz des électrons uniforme (homogène) pour décrire
l'énergie d'échange-corrélation.
L'idée est de remplacer n par n(r) en
considérant que le gaz d'électrons inhomogènes localement
comme homogène. Ce qui revient alors à négliger les effets
des variations de la densité. Alors le terme
d'échange-corrélation ne dépend que de la valeur locale
n(r). L'expression de l'énergie d'échange-corrélation
s'écrit alors :
Equation 1.19 ??????
?????? [??] = ? ??(??) ??????(??)????
Le terme åxc désigne l'énergie d'une seule
particule de gaz d'électron et peut être séparé en
deux termes :
Chapitre I Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
15 | Page
Chapitre I Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
Equation 1.20 e???? = e ?? + e??
Dans le cas si on tient compte de spin, l'approximation LDA
n'est pas valable, alors on utilise l'approximation de LSDA (en anglais Local
Spin Density Approximation).
2.8.2 Approximation de gradient
généralisée GGA :
L'approximation de gradient généralisée
GGA (en anglais Gradient Generalized Approximation) est une deuxième
génération de fonctionnelle utilisée pour décrire
le potentiel d'échange-corrélation [18]. Cette approximation
permet de décrire les systèmes réels inhomogènes et
de mettre en face les contradictions requis avec les résultats
expérimentales trouvées par la LDA. Les calculs LDA ne sont pas
précis, un taux d'erreur sous-estimée comparable est requis en
qui provient de terme d'échange, alors que le terme de
corrélation est toujours surestimé. Cette approximation est
présente pour corriger et diminuer ces taux d'erreurs. La fonctionnelle
GGA tient compte de gradient d'intensité
?ñ(r), alors on peut écrire :
Equation 1.21 ????????? ???[??] = ? ??(??)??[??(??),
????(??)]????
Les deux approximations LDA et GGA sont des approximations de
champ moyen, car elles traitent les interactions
d'échange-corrélation comme l'interaction entre une particule et
un bain des toutes les autres particules.
2.8.3 Approximation de gradient
généralisée GGA-PBE et GGA+U
L'approximation de Perdew-Burke-Ernzerhof PBE [19] est une
nouvelle génération de l'approximation GGA qui permet aussi de
décrire mieux le potentiel d'échange-corrélation.
L'énergie d'échange peut être déterminée
comme une intégrale de densité d'échange.
Equation 1.22 e?? ?????? (??(????);
s(????)) = e??
??????(??(????))??Ç????(s(????))
16 | Page
8
Equation 1.23 ???? ??????(??) = -???? ? ????????(??,
??)
??
Equation 1.24 ??????????(????) = ? ???? ??
(????)?????? ????(??(????); ??(????))
Les valeurs mal estimés trouvés par
l'approximation GGA-PBE peut être corrigés par le terme Hubbard U.
Cette méthode s'appelle GGA+U et destinée pour traiter les
électrons externes 3d et 4F de l'élément de transition. Ce
terme Hubbard peut prendre plusieurs valeurs (1, 2, 8, 8.5...).
2.9 Fonctionnelle hydride B3LYP :
Le traitement de l'échange est exact mais celui de la
corrélation est partiellement omis. La fonctionnelle hybride B3LYP
(Becke, 3 paramétres, Lee, Young, Pan) a été
proposée par Stephens en 1994 [20]. Elle possède deux bases, base
6-31G et la base 6-311G et elle utilise 3 paramètres pour
mélanger dans la exacte HF les xc et LYP.
Equation 1.25 ???????????????? = ?????????? + ????
(?????? ?? - ??????????) + ???? (?????? ???? - ??????????) + ?????????? +
????(???? ?????? - ???? ??????)
Avec a0 = 0.20 , ax = 0.72 et ac
= 0.81
Cette approximation permet aussi d'éviter la
surestimation de l'énergie totale pour le terme
d'échange-corrélation. Elle permet d'approximer la valeur de
l'énergie d'échange-corrélation pour décrire
l'équilibre de la géométrie (énergie d'absorption,
énergie d'excitation...).
Elle utilise l'ajout d'une fraction de corrélation
d'échange d'HF pour annuler une partie de l'auto-interaction [21] (en
anglais Self Interaction).
Elle est caractérisé par deux paramètres
de réseau a et c et un autre paramètre U associé avec la
position atomique.
Chapitre I Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
17 | Page
2.10 Résolution des équations de Kohn et
Sham : Les orbitales de Kohn-Sham (KS) :
Equation 1.26 ????(??,??) = ? ??????
????(??,??)
Avec Cji : Sont les coefficients de
développement.
La résolution des équations nous oblige de
savoir les coefficients Cji. Alors il faut un cycle
d'itérations auto-cohérent en injectons la densité de
charge initiale Pin pour diagonaliser
l'équation séculaire.
Equation 1.27 H-????S=0
Avec H : matrice Hamiltonien et S : matrice de recouvrement.
La nouvelle densité de charge out Pout
est constituée par les vecteurs propres de cette
équation en utilisant la densité de charge totale obtenue par une
sommation sur tous les orbitales occupés. Si l'on n'obtient pas la
convergence des calculs on mélange les densités Pin
et Pout. On
répète ce cycle jusqu'à on atteint la convergence,
jusqu'à ce que la nouvelle densité électronique soit
égale ou très proche de la précédente.
Equation 1.28 ????????+?? = (?? - ??)???????? + ??
????????'
Avec ?? : Paramètre de mixage et
i : présente l'iéme itération
Chapitre I Généralités sur le
TiO2 et la théorie de la fonctionnelle de la
densité
18 | Page
2.11 Cycle auto-cohérant de la résolution des
équations de Kohn et Sham :
|
Structure cristalline
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Calcul atomique H? = E? ñ Atomique
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Superposition des ñ
atomique

Si non
Calcul de potentiel ; équation de poisson
Résolution des équations de
Kohn et Sham,
calcul des
orbitales ? i
Calcul de ñout
ñinit = ñout Convergence

Arrêter
FIGURE 1.5 - Cycle
auto-cohérant de la résolution des équations de Kohn et
Sham
Conclusion :
Ce premier chapitre est l'état de l'art de notre
travail, nous avons présenté quelques
généralités sur le dioxyde de titane et leurs applications
dans plusieurs domaines. Par la suite, nous avons énoncé la
théorie de la fonctionnelle de la densité DFT.
Chapitre II
Méthodes de calcul et matériels
I. Chapitre II Méthodes de calcul et
matériels
20 | Page
Technique d'élaboration des couches minces
de TiO2 : Spray pyrolyse
1.1 Le Spray pyrolyse :
Le spray pyrolyse est une technique très facile
d'élaboration des couches minces. Le mot spray en anglais indique le jet
d'un liquide (parfum, déodorant ...) projeté par des fines
gouttelettes par pulvérisation. Le mot pyrolyse indique un chauffage du
substrat. Le dépôt de films minces uniformes et de haute
qualité peut être divisé en deux grandes catégories
en fonction de la nature du processus de dépôt. Les
méthodes physiques comprennent le dépôt physique en phase
vapeur (PVD), l'ablation au laser, l'épitaxie par jet
moléculaire.
Les méthodes chimiques peuvent être
divisées en deux groupes : les méthodes de dépôt en
phase gazeuse et les techniques de mise en solution. Les méthodes en
phase gazeuse sont le dépôt chimique en phase vapeur (CVD) et
l'épitaxie en couches atomiques, tandis que les méthodes de
pyrolyse par pulvérisation, de revêtement sol-gel, de spin et de
trempage utilisent des solutions de précurseurs.
1.2 Système expérimental de Spray pyrolyse
:
La présente étude vise à préparer
des films minces de TiO2 en utilisant le procédé de
dépôt par pyrolyse par pulvérisation économique. La
technique de pyrolyse par pulvérisation est particulièrement
intéressante, non seulement parce qu'elle donne naissance à des
films de qualité relativement élevée, mais aussi parce
qu'elle est simple, peu coûteuse, peu coûteuse et qu'elle permet de
contrôler de nombreux paramètres tels que la température du
substrat, la composition du précurseur du mouvement de la buse de
pulvérisation. Ces paramètres peuvent influer sur la structure de
l'échantillon préparé et, à son tour, sur ses
propriétés physiques, notamment la structure cristalline, la
taille des particules, la forme et la cristallinité. Aujourd'hui, le
procédé de technique de pyrolyse par pulvérisation est
utilisé pour préparer différents types de
matériaux, qui sont utilisés dans diverses technologies, telles
que les dispositifs optoélectroniques, les cellules solaires et les
systèmes de capteurs.
Le système de dépôt par pyrolyse par
pulvérisation est représenté schématiquement sur la
figure (2.1), il contient essentiellement une buse de pulvérisation
ronde, une pompe et une plaque chauffante.
Chapitre II Méthodes de calcul et
matériels
21 | Page
1.2.1 Le spray nozzle :
La solution à pulvériser est
pulvérisée sur le substrat chauffé à travers une
buse à jet rond dont l'orifice a un diamètre de 0,5 mm et
contient un conduit pour le liquide et un autre pour le transporteur de gaz. La
buse est montée sur un système de balayage XY lui permettant de
couvrir la totalité de la surface active de la plaque chauffante par
passages réguliers et successifs du jet. Le vecteur de gaz disperse la
solution en gouttelettes pour former l'aérosol qui recouvre le substrat
à une certaine vitesse comme un jet conique. Le gaz
sélectionné est l'azote (N2) qui est inactif lors de la formation
du composé par des réactions chimiques et qui évite les
chimisorptions de l'oxygène lors de la formation des couches.
Le choix de la vitesse est déterminé par la
distance entre le substrat et la buse. Ce paramètre influence en effet
la taille des gouttelettes, puis la qualité du film formé sur le
substrat chauffé. Afin de réaliser une pulvérisation
uniforme sur toute la surface du substrat, la distance de la buse du substrat
est égale à 30 cm.
1.2.2 Pompe :
Le débit de la solution à pulvériser est
contrôlé par une pompe aspirante (ISMATEC). Le choix du
débit de pulvérisation résulte d'un compromis entre
vitesse de croissance des couches la plus rapide possible, bonne
cristallinité et orientation privilégiée des grains. Pour
cela, nous avons réglé les débits de liquide et de gaz aux
valeurs respectives de
4mL.mn -1 et
4L.mn -1.
1.2.3 Plaque chauffante :
La plaque chauffante utilisée est un bloc de fonte
grise, de dimensions 12 x 10 x 3 cm3,
parallélépipédique, fabriqué pour assurer un
contact le plus parfait possible avec le substrat. A l'intérieur de
cette plaque, trois éléments chauffants sont connectés en
parallèle l'un à côté de l'autre afin d'avoir une
température suffisamment homogène sur toute la surface de la
plaque. Le pouvoir de chacun la résistance est de 100 watts.
Pour assurer la stabilité thermique de la plaque, un
type d'alimentation « PYROLABO » muni d'un thermocouple de
contrôle (régulateur de température) qui est introduit dans
le bloc.
Chapitre II Méthodes de calcul et
matériels
22 | P a g e
La température du substrat Ts pendant la
pulvérisation est contrôlée à l'aide d'un
deuxième thermocouple, dont le fin est intégrée dans une
très petite quantité de étain fondu (pureté 99%,
température de fusion = 232 O C) appliqué à un
contrôle du substrat température.

FIGURE 2.1 - Schéma
général d'un procédé de dépôt par
pyrolyse par pulvérisation
II. Logiciel Materials Studio :
2.1 Présentation :
Materials Studio [22] est un logiciel de simulation
numérique commercialisé par Accelrys Inc (c) qui permet
l'étude des propriétés physico-chimiques des
matériaux. Il capable de construire et manipuler en 3D les
modèles graphiques des molécules, matériaux amorphes, les
surfaces...
Chapitre II Méthodes de calcul et
matériels
FIGURE 2.2 - Logiciel Materials
Studio
Le calcul est lancé à la faculté des
sciences de Gafsa. Ce calcul est très puissant, nous avons
travaillé sur une station serveur doté de 256 Go mémoire
RAM et 4 processeurs (AMD Opterom (tm) Processor 6328) de 3.2 GHz
fréquence de traitement. Afin d'installer tous les logiciels
nécessaires, il faut savoir que la station doit être toujours
allumé pour ne pas perdre le calcul. Durant le calcul la
température de la station vat augmenter progressivement. Alors nous
sommes obligés d'allumer un systéme de climatisation qui va
être mis en marche en parallèle avec la station. D'une autre part,
il faut notez bien que le calcul d'être terminé sans échec,
chaque rupture ou verrouillage de la station automatiquement va cesser le
calcul. Alors il faut contrôler le calcul même si on n'est pas sur
le champ. A l'aide d'une simple application TEAM VIEWER, nous pouvons
contrôler le calcul et faire tout ce qu'on veut à distance
à l'intermédiaire de l'adresse IP et le mot de passe de la
station par un PC ou sur l'Android par un smart phone.
23 | P a g e
(A) (B)
FIGURE 2.3 - (A) Station de
calcul, (B) Logiciel Team Viewer
Chapitre II Méthodes de calcul et
matériels
24 | P a g e
2.2 Code CASTEP :
2.2.1 Présentation :
CASTEP (en anglais Cambridge Serial Total Energy Package) [23]
est un logiciel qui utilise la théorie de la fonctionnelle de la
densité. C'est un code inclut dans Materials Studio et basé sur
la mécanique quantique pour calculer l'énergie totale de
l'état fondamental. Pour la résolution numérique, CASTEP
nécessite une décomposition des orbitales de Kohn-Sham qui sont
pratiquement des ondes planes. Il utilise les méthodes de
Pseudo-Potentiels PP et de ondes planes PW [24] pour résoudre les
équations de Kohn-Sham. Le pseudo-potentiel ultra-doux de Vanderbilt est
utilisé pour décrire l'interaction entre les électrons de
coeur et les électrons de valence [25]. D'une autre part, CASTEP utilise
les approximations LDA et GGA pour décrire l'interaction
d'échange-corrélation.
Pour calculer l'énergie totale, il utilise une
intégration des K-points dans la première zone de Brillouin ZB,
des pseudo-potentiels, des ondes planes et des supermailles. Il remplace les
électrons de coeur [26] par des potentiels agissant uniquement sur les
électrons de valence. Cette approximation est connue sous le nom
approximation Frozen-Core. L'exécution doit avoir deux fichiers un
fichier de sortie et un fichier binaire formaté qui contient les
coefficients de la fonction d'onde et la densité de charge.

FIGURE 2.4 - Code CASTEP dans
Materials Studio
Chapitre II Méthodes de calcul et
matériels
25 | P a g e
2.2.2 L'optimisation de la géométrie avec
CASTEP :
L'optimisation de la géométrie est une tâche
très importante avant le calcul. Nous ne pouvons pas calculer tels
propriétés que lorsque la structure du matériau soit
stable.
Alors avant d'entamer le calcul, CASTEP contient une tâche
d'optimisation de la structure qui permet d'obtenir une structure stable.

FIGURE 2.5 - L'optimisation de la
géométrie avec CASTEP
2.2.3 Théorème de Bloch :
Certains matériaux contiennent un grand nombre des
atomes et des électrons, ce qui rend difficile la résolution.
Dans ce cadre, pour les matériaux qui présentent
une périodicité, on peut simplifier la résolution. On
passe d'un systéme infinie d'équations à un systéme
finie pour un nombre infinie des points K.
Alors le théorème de Bloch donne une autre
formulation de la fonction d'onde.
Equation 2.1 ???? ??(??) = ??????????????
??(??)
Chapitre II Méthodes de calcul et
matériels
2.2.4 Echantillonnage de la Zone de Brillouin ZB :
L'énergie totale de système ne peut être
calculée que par l'intégration de la zone de Brillouin ZB. Pour
la précision de l'intégration il faut échantillonner la ZB
la plus finement possible. Dans ce cadre, Monkhorst et Pack [27] ont
proposés une méthode d'échantillonnage la plus
répandue qui permet de simplifier le premier ZB et d'obtenir une grille
uniforme de points k de dimension choisie.
2.2.5 L'énergie de coupure et super cellules
:
L'énergie de coupure Ecutof est
l'énergie de l'onde plane dont la fréquence la plus
élevée, qui doit être fixée pour développer
les orbitales Kohn-Sham. Elle contrôle la convergence de calcul et de
base pour représenter la fonction d'onde nous indique la coupure sur le
nombre des fonctions d'ondes planes utilisées comme fonctions.
Les super cellules sont utilisées pour décrire
les surfaces. Parfois pour qu'on désire améliorer la vitesse de
calcul, on peut faire la symétrie on ajoute la super cellule pour
accélérer les calculs longs. On introduit un vide artificiel en
considérons un arrangement d'une structure quel conque (CFC, CC...) qui
contient tous les atomes. Les structures sphériques sont construites en
remplaçant un à un les atomes de matériau hôte qui
vont être fortement liés comprimées. On peut
générer plusieurs types de super cellules telles que (2 X
2 X 1), (2 X 2 X
2), (1 X 2 X 1)... Les figures
ci-dessous montrent deux types de super cellules :

26 | Page
(A) (B)
FIGURE 2.6 - Super cellule de la
phase anatase : (A) Super cellule (2 X 2
X 1) et (B) Super
cellule (3 X
3 X 1).
Chapitre II Méthodes de calcul et
matériels
27 | Page
III. Dynamique Moléculaire DM :
3.1 Introduction :
Parfois certaines expériences sont impossibles de les
faire comme à l'intérieur des étoiles, la
dangerosité qui provoque le risque d'explosion... Le physicien
américain «Richard P. Feynman» a dit que « That
all things are made of atoms, and that everything that living things do can be
understood in terms of the jigglings and wigglings of
atoms.» Le but est très clair, on cherche
à comprendre comment les atomes et les molécules se
déplacent et vibrent...
Ce pour cela la simulation est la seul clé pour faire
ces expériences facilement sans risque et sans aucun doute. Ces
simulations sont parfois appelés « expériences
théoriques » cause qu'ils peuvent effectuer des expériences
difficiles à faire.
Dans ce cadre, nous sommes obligés de faire une telle
simulation pour mettre en faces ces problèmes et gagner le temps de
plus. Généralement il existe deux types de simulation, une
simulation qui utilise l'approche quantique et une autre utilise l'approche
classique dite DM. La dynamique moléculaire DM (en anglais Molecular
Dynamics Simulation) est apparue en 1930 [28], mais elle s'est
développée vers les années soixante. Cette théorie
est basée sur l'approximation de BO [29] ou les électrons sont
plus rapides que les noyaux.
La dynamique moléculaire est un outil très
important pour comprendre les propriétés statistiques et
dynamiques, elle décrit les modes vibrationnelles des réseaux
cristallins en calculant les vitesses et les positions en fonction de temps.
Cette méthode de calcul est incluse dans le code CASTEP dans Materials
Studio. On s'intéresse à la description des atomes et les
interactions moléculaires en vue de calculer l'énergie totale de
système. La propagation de système dans l'espace des phases se
fait en appliquant les équations classiques du mouvement. Cette
théorie permet de suivre l'évolution temporelle d'un ensemble de
N atomes par l'intégration numérique pas à pas des
équations du mouvement classique pour comprendre les
propriétés des assemblages des molécules en termes de
structure et les interactions microscopiques.
L'idée est si on connait le potentiel d'interaction
entre les atomes ou molécules, on peut utiliser la relation fondamentale
de la dynamique RFD pour suivre l'évolution en fonction de temps des
positions de chaque atome en intégrant numériquement les
équations du mouvement.
Chapitre II Méthodes de calcul et
matériels
28 | P a g e
En mécaniques classique, chaque particule est
définit comme [30] une masse ponctuelle soumise à une interaction
mutuelle. Chaque particule définit par trois degré de
liberté relative aux positions et trois relatives aux impulsions. Le
système est donc déterminé par six degrés de
liberté.
3.2 Les équations du mouvement :
Un système contient N particules
considérées comme étant des masses ponctuelles de masse m,
ayant K énergie cinétique et V énergie potentielle alors
les équations du mouvement sont :
Equation 2.2 ????? = ????
??????
Equation 2.1 ????? = - ????
??????
Avec H = K + V
On peut exprimer le mouvement des atomes grâce à
l'équation fondamentale de la dynamique :
|
RFD: ? ???? = ???????? = ????
|
????????
Equation 2.4
??????
|
Avec : Fi la force totale exercée mi : La masse atomique
i
d2ri
???? = dt2 : L'accélération
Cette force s'écrit :
Equation 2.5 ???? = -
????V ou ???? =
????
?????? + ??????? ??? + ????
??????
Chapitre II Méthodes de calcul et
matériels
29 | P a g e
Pour décrire le systéme à N atomes en
interaction la première équation (2.4) conduit à
résoudre 3N l'équation différentielle à 2
ordres.
3.3 Algorithmes d'intégration numérique :
La résolution des équations du mouvement se fait de
manière discrète en utilisant la méthode de
différence finie. Une fois si on connait la vitesse et
l'accélération de la particule, alors on peut calculer V et U
pour t+?t avec ?t est le pas d'intégration.
On est besoin des algorithmes rapides et qui demandent un cout
informatique faible et doit permettre l'utilisation de grands pas
d'intégration ?t et satisfaire les lois de conservation d'énergie
et de moment.
3.3.1 Algorithme Velocity Verlet :
L'algorithme Verlet a été
développé par le physicien français Loup Verlet en 1967
[31], basé sur l'approche des différences finies pour
résoudre les équations du mouvement. Il offre une bonne
stabilité de calcul que la méthode d'Euler. La précision
de ce algorithme est donné par ?t4Nt et le temps
maximale écoulé par la simulation est donné par ?t
Nt. Pour intégrer numériquement les équations
différentielles, il est nécessaire de les discrétiser en
temps.
Comme il est important que l'énergie de systéme
soit conservée au cours de temps. L'algorithme de Verlet s'écrit
sous forme de vitesse présentée par deux équations
à l'aide d'un développement de Taylor :
Equation 2.6 (r+??? ) =
r(t) + v(t)
?t+ ??(??(??))
???? ????? + ??????
??????
Equation 2.7 (r-??? ) =
r(t) - v(t)
??t+ ??(??(??))
???? ????? - ??????
??????
L'équation (II.6) + (II.7) donnent :
Equation 2.8 (?? + ???) +
(r-???) = 2r(t) +
??((????))
???? ?t?? + ?
30 | Page
Chapitre II
|
|
|
|
|
Méthodes de calcul et
matériels
|
|
La position :
|
???? (??
|
+ ???)
|
=
|
???? (??)
|
+ ???? (??) + ???? ???? (??)?????
|
Equation 2.9
|
|
La vitesse :
|
????(??
|
+???)
|
=
|
????(??)
|
+ ?? ?? ???[????(??) + ????(?? + ???)]
|
Equation 2.10
|
Maintenant on connait la position et la vitesse alors nous
pouvons obtenir l'état de système au temps (t + ?t) + ?t.
L'intégration complète des équations du mouvement suit un
schéma à 4 étapes :
· Etape 1 : Evaluer les forces Fi(t) à partir des
positions ri(t).
· Etape 2 : Calcul des nouvelles positions ri(t + ?t).
· Etape 3 : Evaluation des forces Fi (t+?t) à
partir des positions ri (t+?t).
· Etape 4 : Calcul des vitesses Vi
(t+?t).
Le pas du temps utilisé pour la simulation étant
finie, cela peut induire des erreurs sur le calcul des postions et vitesses.
Ces erreurs augmentent rapidement et le calcul peut diverger.
3.3.2 La symétrie par renversement du temps de
Verlet :
Pour tester le degré des erreurs d'arrondie, Verlet
satisfait bien cette symétrie, en
changeant +?t par - ?t l'équation du mouvement reste
inchangée et donc la trajectoire de la DM revient sur ces pas.
3.3.3 Algorithme de saute-mouton :
L'algorithme de saute-mouton (en anglais Leap-Frog) dérive
de l'algorithme originel Verlet qui permet d'évaluer les vitesses
à des temps semi-entier de ?t pour t - ?t2 et pour t + ?t . Cet
algorithme saute de V?? (t - ?t 2 ) a V?? (t +
?t
2 ) sans utiliser de V??(t) ,
d'où son le « saute-
2
|
mouton ».
Equation 2.11 Equation 2.12
|
??
????
|
(??
(??
|
-
+
|
???
|
=
=
|
??(??)-????(??- ?????)
|
|
|
??)
???
|
???
????(??+???)-????(??)
|
|
?? )
|
???
|
Chapitre II Méthodes de calcul et
matériels
31 | Page
On tire alors : ????(?? + ???) = ????(??) + ????
(?? + ???
?? ) ??? Equation 2.13
La vitesse s'écrit : ???? (?? + ??
???) = ????(?? - ???
?? ) + ??(??)
?? ??? Equation 2.14

FIGURE 2.7 - Principe de
l'algorithme du Verlet-Leap-Frog [32]
3.4 Conditions périodiques CP :
Pour rendre le systéme pseudo-infinie, la simulation
nécessite que la taille de système doit être
inférieur à l'échelle macroscopique et le nombre des
particules doit être inférieur au nombre d'Avogadro. Par ailleurs,
on se limite à la taille du système à quelques
molécules introduirait des effets de bords inadmissibles. Dans ce cadre
il faut qu'une portion de système réel placé dans un
espace finie appelé boite de simulation.
3.5 Boite de simulation :
La boite de simulation dépend des plusieurs
paramètres tel que longueur de côté L, forme, nombre
d'atomes. Le volume de la boite de simulation est V = L3 = LxLYLZ et
sa taille de la est de 20 à 100Å. La forme de la boite peut varier
suivant les systèmes étudiés. Pour les liquides et les
amorphes, on adopte la forme cubique ; les CP génèrent un milieu
isotrope.
Chapitre II Méthodes de calcul et
matériels
32 | P a g e
Pour une structure cristalline, les CP doivent refléter
la symétrie transrationnelle du cristal et les CP sont donc
appliqués dans le système d'axes cristallographique.
Dans la boite à 2D [33] il y 8 boîtes image. En
3D, il y a 26 boîtes image. Tout mouvement d'une particule dans la
boîte primitive correspond un mouvement fictif de toutes ses images
périodiques. Lorsqu'une particule sort de la boîte origine par une
des faces, l'image de cette particule entre dans la boîte par la face
opposée. Cela permet de garder le nombre de particules constant dans la
boîte, et la masse, l'énergie et le moment cinétique sont
conservés au cours de la simulation. Les interactions interatomiques
sont à courte portée, on prend compte que la distance la plus
courte, alors Les CP ne peuvent pas être utilisé dans le cas des
interactions à longue portée.

FIGURE 2.8 -
Représentation schématique de la duplication de la boite de
simulation à deux dimensions. En utilisant les conditions aux limites
périodiques, lorsqu'une particule se déplace et quitte la boite
principale, ses images dans les cellules voisines se déplacent de la
même façon.
3.6 Potentiels :
Le comportement de système est défini par les
interactions entre les atomes de système d'où les
résultats de la simulation dépendent des potentiels effectifs. Ce
potentiel a pour rôle de reproduire l'énergie potentielle V qui
dépend de 3N coordonnées des atomes.
Dans le modèle de ce potentiel effectif ce potentielle
s'écrit :
Chapitre II Méthodes de calcul et
matériels
33 | Page
Equation 2.15 V (????, ... ????) ????
+ ? ?? ???? (????) + ??? ???>?? ???? (????, ????) +
??? ???>?? ???>??>?? ???? (????, ????, ????)
3.7 Calcul d'énergie et des forces :
Le calcul est basé sur la méthode de sommation
d'Ewald. Dans une boite cubique de côté L, un système
à N particules chaque atome ayant une position ri et une charge qi.
Equation 2.16 ??? ???? = ??
L'énergie totale de système s'écrit alors
:
?? ????????
8 ??
Equation 2.17 U = ? ? ?
??
??=?? ??=?? ??=??
?? |????-(????+????)|
U s'écrit aussi : U = ???? +
???? - ???? Equation 2.18
UD : Énergie d'espace directe UR :
énergie d'espace réciproque US : Terme de
correction
3.8 Thermostat :
Le thermostat joue un rôle très important durant
toute la simulation, il permet de maintenir la température de
systéme constante. On trouve plusieurs thermostats tels que le
thermostat de Nosé-Hoover, Barostat de Nosé-Hoover, Langevin
...
3.9 Déroulement de la simulation DM :
En absence de contraintes extérieures, l'énergie
de système reste conservée durant toute la simulation, alors
l'ensemble micro-canonique NPE [34] est considéré comme
l'ensemble naturel de la simulation. La température et la pression ne
peuvent pas être contrôles, alors il faut considérer
d'autres ensembles tels que NVT, NPH, NPT...
Chapitre II Méthodes de calcul et
matériels
34 | P a g e
La simulation est réalisée sur des systèmes
finis, de ce fait la petitesse des particules un ensemble statistique
adéquat NPE, NVT... Les équations Newtoniennes impliquant ensuite
une conservation de l'énergie au cours de temps.
Les surfaces doivent jouer un rôle important sur les
propriétés simulées. Alors les effets de bord ou de
surface ne sont pas négligeables, nous s'intéressons à la
structure des matériaux infinis c'est-à-dire rendre le
systéme pseudo-infinie.
3.9.1 Les conditions initiales :
? Afin de définir une boite de simulation dans laquelle
les particules sont placées, il faut fixer la position de départ
des molécules et en les donnant une vitesse initiale. ? L'ensemble
statistique soit NVT, soit NPH, soit NPT, soit NPE...
? Il y'a plusieurs ensembles statistiques de simulation nous les
montrons ci-dessous.
Variables d'état Ensemble
statistique
E constant, V constant NVE (l'équilibre de
systéme)
P constant, T constant NPT
P constant, H constant NPH
T constant, V constant NVT (chauffage de
systéme)
TABLEAU 2.1 - Ensembles
statistiques
3.9.2 Equilibration :
Cette étape consiste à amener le système
d'une configuration initiale à une configuration représentative
de systéme à partir de laquelle on peut effectuer un
échantillonnage respectant la condition de micro
réversibilité et de modéliser les systèmes
désordonnés. A ce stade, le système oublie son état
initial et les caractéristiques macroscopiques atteintes leur valeur
équilibre : c'est la relaxation.
Chapitre II Méthodes de calcul et
matériels
35 | P a g e
3.9.3 Production :
Afin de calculer les propriétés macroscopiques,
cette étape consiste à sauvegarder les caractéristiques de
systéme pendant un grand nombre de configuration. La trajectoire de
l'espace de phase (position, vitesse et accélération) est
sauvegardé à intervalle régulier, puis utilisé pour
calculer les observables de système.
Conclusion :
Dans ce chapitre nous avons présenté tous les
méthodes de simulation et les matériels utilisés. En
premier, nous avons présenté la technique « Spray Pyrolyse
» pour l'élaboration des couches minces de dioxyde de titane TiO2.
Par la suite, nous avons explicité le logiciel de simulation «
Materials Studio » ainsi le code serveur « CASTEP ».
D'une autre part, nous avons terminé ce chapitre par la
méthode de simulation « Dynamique Moléculaire Simulation
».
36 | Page
Chapitre III
Résultats et discussions
Chapitre III Résultats et
discussions
37 | Page
3.1 Le détail de calcul :
Dans notre travail, nous avons effectué le calcul des
premiers principes au sein de la faculté des sciences de Gafsa. Ce
calcul ab-initio est basé sur la théorie de la fonctionnelle de
la densité DFT. En utilisant la méthode d'ondes planes et
pseudo-potentielles implémentées dans le code CASTEP de Materials
Studio.
La première étape consiste à optimiser le
nombre de points spéciaux « k » pour le maillage de la
première zone de Brillouin partir d'une mèche fine.
Les énergies d'interactions d'échanges et de
corrélations sont évaluées par les approximations de LDA,
GGA-PBE, PBEsol, GGA + U et la fonctionnelle B3LYP.
Tous les travaux sont effectués avec le dioxyde de
titane anatase. Cette phase est de symétrie tétragonal, et
décrit par le groupe d'espace I41/amd. Les paramétres
de maille sont
respectivement les suivants : a = b = 3.780 A, c = 9.510 A et
C/?? = 2.51 > 1.
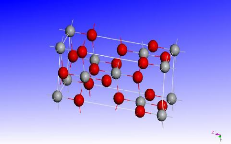
FIGURE 3.1 - Structure
cristalline du dioxyde de titane anatase
3.2 Optimisation de la géométrie :
La tâche d'optimisation de la géométrie
est le point de départ de tous les calculs, quel que soit le calcul et
quel que soit le code utilisé. Comme on a dit dans le chapitre
précèdent, elle est très importante pour que la structure
soit parfaitement stable pour qu'on puisse calculer les
propriétés recherchés.
Nous avons utilisé le code CASTEP dans Materials Studio
pour faire l'optimisation de TiO2 anatase et on répète
l'optimisation jusqu'on obtient la convergence.
Chapitre III Résultats et
discussions
38 | P a g e
Dans le but de connaitre la valeur de l'énergie de
coupure nécessaire pour notre calcul nous avons fait un petit
diagnostic. Nous avons fait plusieurs calculs par plusieurs valeurs (100, 200,
300, 450, 500, 550, 600 eV) d'énergie de coupure pour une bonne
convergence.
Le tableau suivant montre les différentes valeurs de
convergence :
|
Energie de
|
|
|
|
|
|
|
|
|
coupure en (eV)
|
100
|
200
|
300
|
450
|
500
|
550
|
600
|
|
Energie de convergence en (eV)
|
-384.194
|
-423.261
|
-429.213
|
-429.579
|
-429.598
|
-429.599
|
-429.634
|
TABLEAU 3.1 - Variation de la
convergence en fonction des différentes valeurs d'énergie
de coupure

FIGURE 3.2 - Variation de la
convergence en fonction de l'énergie de coupure
Chapitre III Résultats et
discussions
39 | P a g e
La figure (3.2) montre que la convergence à partir de -
429 eV reste la même valeur pour les énergies de coupures 450,
500, 550 et 600 eV. On comprend alors que la valeur adéquate de
l'énergie de coupure est 450 eV.
Dans ce cadre, l'optimisation de la géométrie
est réalisée sur une base d'ondes planes avec une énergie
de coupure de de 450 eV. Nous avons utilisé les potentiels Ultrasoft et
le potentiel a norme conservé pour décrire l'interaction entre
les électrons de coeur et les ions du coeur.
La méthode de Monkhorst - Pack avec des k - points
(3×3×2) est aussi utilisé pour simplifier la zone de
Brillouin. Pour décrire la fonctionnelle
d'échange-corrélation Exc., nous avons utilisé la
fonctionnelle GGA-BPE.
Nous avons obtenu la convergence après 7
itérations, avec une énergie 2.98.10-5 ev/atom, et un
maximum de force ionique 5.93.10-3 eV/Å. Le maximum de
déplacement entre les cycles d'itérations est
8.030.10-4 Å et le maximum de pression 8.669.10-2
GPa. Les figures (3.3) ci-dessous montrent les différentes étapes
d'optimisation, la convergence et l'optimisation en fonction de
l'énergie.

(A)

(B) (C)
FIGURE 3.3 - (A) Structure avant
l'optimisation, (B) et (C) : Optimisation de la géométrie
du TiO2

Chapitre III Résultats et
discussions
FIGURE 3.4 - La convergence
d'optimisation avec CASTEP
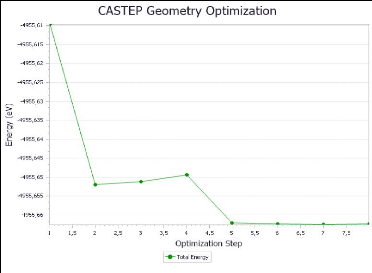
40 | P a g e
FIGURE 3.5 - L'optimisation de la
géométrie du TiO2 anatase en fonction d'énergie
Chapitre III Résultats et
discussions
41 | P a g e
3.3 Spectre Raman :
La spectroscopie Raman est une méthode de
caractérisation des vibrations de réseaux cristallins. On cherche
toujours à observer les modes de vibration du réseau cristallin
par l'identification des différentes modes de vibrations. Le calcul est
basé sur la théorie de la DFT. Pour décrire l'interaction
d'échange - corrélation, nous avons utilisé la
fonctionnelle hybride B3LYP.
|
a
|
b
|
C
|
Alpha
|
Beta
|
Gamma
|
|
3.7617561 Å
|
3.7617561 Å
|
3.7617561 Å
|
90.00000
|
90.00000
|
90.00000
|
(a)
|
x
|
Y
|
Z
|
O ( 8 )
|
4.656804416270E-17
|
2.500000000000E-01
|
1.721700410720E-01
|
Ti ( 22 )
|
5.000000000000E-01
|
-2.500000000000E-01
|
-1.250000000000E-01
|
|
(b)
TABLEAU 3.2 - (a)
Paramétres de maille de l'anatase, (b) Coordonnées
cartésiens
Les figures (3.6) et (3.7) montre respectivement les spectres
Raman du TiO2 anatase expérimentale et théorique obtenue par
DFT/B3LYP.

Figure 3.6 - Spectres Raman
expérimentale du TiO2 anatase

Chapitre III Résultats et
discussions
42 | P a g e
Figure 3.7 - Spectres Raman du
TiO2 anatase DFT/B3LYP
La maille de l'anatase est tétragonal appartient au groupe
d'espace D4h
19 (I41 ?? ????). La maille
primitive de l'anatase possède deux groupements TiO2 (donc
6 atomes) par maille ce qui conduit a 15 modes de vibrations possibles (3 x 6 -
3).
La théorie des groupes donne la représentation
irréductible suivante pour les vibrations optiques de l'anatase [35]:
Equation 3.1 ???????? + ?????? ?? + ???????? + ????????
+ ?????? + ??????
Les modes ??????, ?????? et ???? sont actifs en spectroscopie
Raman et les modes ?????? et ???? sont actifs en spectroscopie infrarouge. Par
contre, le mode ?????? est inactif ni en spectroscopie Raman ni en
spectroscopie infrarouge.
Chapitre III Résultats et
discussions
43 | P a g e
En spectroscopie Raman il y a 6 modes de vibrations permis.
Les modes actifs en spectroscopie Raman sont les quatre modes ??????,??????,
?????? et ????, les autres sont actifs en spectroscopie infrarouge et le mode
A2g est silencieux [36].
La figure (3.6) [37] montre 4 pics c'est à dire 6
bandes situées respectivement à 144 cm bande ????, à 398
cm bande ?????? , à 515 cm bande ?????? et a 639 cm bande ????.. Nous
constatons que le premier pic est la raie la plus intense ????. Cette pic
désigne toujours la raie référence parce qu'elle est la
plus intense et la plus fine ce qui facilite l'observation de ses
déplacements.
La troisième raie est un doublé de deux raies 513
?????? et 519 ??????.
On remarque que les pics de spectre Raman obtenue par
DFT/B3LYP (figure (3.7)) sont les mêmes pics obtenus
expérimentalement (figure 3.6)).
Alors on déduit que notre calcul DFT avec la
fonctionnelle hybride B3LYP est en bon accord avec l'expérimentale.
Dans le tableau (3.3) ci-dessous, nous présentons les
modes de vibrations et la fréquence en (nm) associé à
chaque mode.
|
Mode
|
Fréquence (nm)
|
|
Eg
|
144
|
|
B1g
|
398
|
|
A1g
|
514
|
|
B1g
|
519
|
|
Eg
|
639
|
TABLEAU 3.3 - Fréquences des
différentes bandes Raman de la phase anatase du TiO2 [38]
Pour chaque mode de vibration, nous présentons
ci-dessous la figure (3.8) les modes de vibrations et de déplacements
des atomes [39].
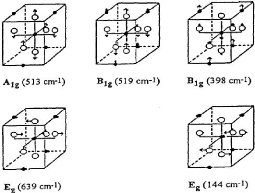
Chapitre III Résultats et
discussions
44 | P a g e
Figure 3.8 - Schéma des
déplacements des atomes pour les différentes modes des
vibrations
3.4 Propriétés électroniques :
3.4.1 Structure des bandes et densité
d'état électronique :
En physique de la matière condensée, la
densité d'état électronique DOS quantifie le nombre
d'états électroniques susceptibles d'être occupés,
et possédant une énergie donnée dans le matériau
considéré. Dans cette partie, nous nous sommes
intéressés aux calculs des structures de bande d'énergie
et les densités d'états électroniques du dioxyde de titane
anatase, en utilisant les paramètres optimisés du réseau.
Les niveaux d'énergie des électrons sont décrits en termes
de fonctions continues En, k (ou En(k)) qui ont la périodicité du
réseau réciproque. Ces fonctions En(k) définissent la
structure de bande.
L'entier n est appelé indice de bande, et k est une
variable continue de l'espace des k, limitée
à la première ZB. Elle s'écrit dans le
cas d'un système de dimension trois :
Equation 3.2 ??(E) = ? ? ??????
???????? ???????? (E -
E????)
Les figures (3.9) et (3.10) montre respectivement les structures
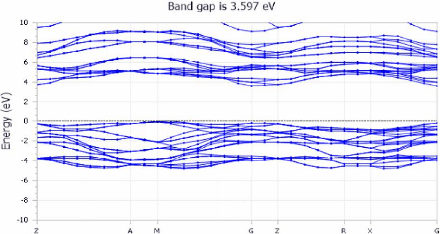
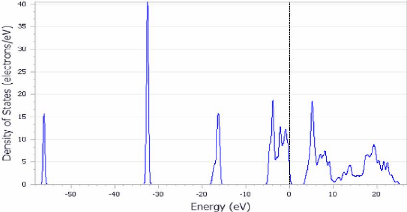
des bandes et les densités électroniques DOS obtenues par DFT.

Chapitre III Résultats et
discussions
45 | P a g e
DFT/LDA

DFT/GGA-PBE
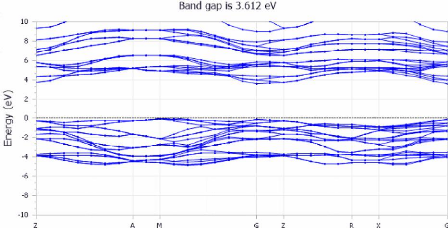
Chapitre III Résultats et
discussions
46 | P a g e
DFT/GGA-PBEsol
DFT/GGA+U
FIGURE 3.9 - Les structures des
bandes du TiO2 anatase

Chapitre III Résultats et
discussions
DFT/LDA

DFT/GGA-PBE
DFT/GGA+U
47 | Page
Chapitre III Résultats et
discussions
48 | P a g e
DFT/GGA-PBESOL
FIGURE 3.10 - Les densités
d'état électronique DOS du TiO2 anatase
Dans le cadre du présent travail, les structures de
bandes électroniques ont été déterminées par
un calcul DFT dans les approximations du gradient
généralisé LDA, GGA-PBE, GGA+U, et GGA- PBEsol.
Pour chaque méthode utilisée nous avons fait
l'optimisation de la géométrie avec le code CASTEP puis on a
effectué le calcul. D'une autre part, c'est très important ; pour
chaque calcul nous avons fait l'optimisation et le calcul des
propriétés avec la mémé approximation.
Les structures des bandes sont représentées dans
l'espace réciproque pour simplifier les calculs. Les calculs ont
été réalisés le long des différents points
et directions de haute symétrie dans la première zone de
Brillouin dans le but de déterminer la valeur de gap
énergétique de ce matériau. Les calculs ont
été effectués en utilisant plusieurs approximations
Les points de haute symétrie : Z--G G--X X--P P--N
N--G.
Le niveau de Fermi Ef est toujours pris comme une origine
d'énergies Ef = 0.
Une possibilité pour contourner les limitations serait
avec GGA+U et d'augmenter le potentiel GGA par la correction d'Hubbard pour
décrire les fortes interactions coulombiennes intra-site
écrantées entre les électrons d.
Cette correction est effectuée avec Ud = 8.5,
nous avons trouvé Eg = 3.597 eV.
Chapitre III Résultats et
discussions
49 | P a g e
A première vue, Les figures (3.9) montrent que : le
composé étudié, est un semi-conducteur à bande
interdite directe (gap direct) puisque le maximum de la bande de valence BV et
le minimum de la bande de conduction BC sont en coïncidence, ils se
trouvent sur la même ligne passant par le point de haute symétrie.
Cette résultat est bien en accord avec l'expérimentale. La bande
de conduction est située au-delà de 3.301 eV et la bande de
valence au-dessous de niveau de Fermi.
Les figures (3.10) montrent aussi que la valeur du DOS de la
phase anatase est importante autour de niveau de Fermi, ce qui prouve la nature
métallique de cette phase.
Méthode LDA GGA-PBE GGA+U GGA-PBESOL EXP Autre
calcul
Bande 3.53 3.607 3.597 3.612 3.6
Mbj 3.01
interdite GW 3.56
TABLEAU 3.4 - Les valeurs de
l'énergie de gap obtenue [40]
D'après les résultats du (tableau 3.3), l'on
arrive à constater que le gap calculé est proche de valeur
théorique de GW et proche aussi de valeur expérimentale.
3.5 Propriétés optiques :
L'étude des propriétés optiques des
semi-conducteurs, est consacrée à la compréhension des
phénomènes d'interaction d'une radiation lumineuse avec la
matière.
Ces propriétés sont très importantes pour
comprendre les propriétés électroniques des
matériaux.
Plusieurs propriétés optiques qu'on peut les
déterminer comme (la fonction diélectrique, la
réflectance, le coefficient d'absorption, la transmittance....).
On doit noter que tous les propriétés optiques
sont calculées avec le code CASTEP avec l'approximation de GGA-PBE et
l'optimisation de la géométrie est aussi effectuée avec ce
même code avec la même approximation.
Chapitre III Résultats et
discussions
50 | Page
3.5.1 Fonction diélectrique :
La réponse optique d'un matériau est souvent
donnée par sa fonction diélectrique. Il s'agit d'une grandeur
complexe possédant une partie réelle et une partie imaginaire.
Cette fonction diélectrique s'écrit sous la forme complexe par la
relation suivante :
Equation 3.3 ?? (w) = ???? (w) + ??????
(w)
Ou ??1 est la partie réelle et
??2 la partie imaginaire.
La figure (3.11) montre la variation de å1(ù) et
å2(ù) de la fonction diélectrique, dont l'énergie
varie de 0 eV jusqu'à 40 eV.

FIGURE 3.11 - Fonction
diélectrique du TiO2 anatase
Nous avons utilisé l'approximation de GGA-PBE (Gradient
Generalized Approximation - Perdew-Burke-Ernzerhof) pour le calcul des
propriétés optiques. A partir de cette courbe nous avons
déterminé la valeur statique de la fonction diélectrique
réelle å1 (0) = 6.65 Nous remarquons que l'intensité
maximale de la partie réelle ??1(w)
coïncide avec une énergie de 4 eV, puis elle commence à
augmenter simultanément avec l'énergie jusqu'à 4 eV puis
elle diminue. La partie imaginaire de la fonction diélectrique est ne
quantité nécessaire et cruciale
Chapitre III Résultats et
discussions
51 | Page
qui indique l'évolution des transitions inter-bandes dans
le semi-conducteur.
Méthode LDA GGA Mbj Exp Notre
calcul
Fonc.diélectrique 6.99 6.75
5.35 5.8 6.65
TABLEAU 3.5 - Fonction
diélectrique du TiO2
Notre calcul est très proche aux autres calculs
théoriques LDA, GGA, Mbj mais un peu loin de valeur
expérimentale.
3.5.2 Coefficient d'absorption :
Le coefficient d'absorption optique est l'un des critères
d'évaluation les plus importants pour les propriétés
optiques. La figure (3.12) montre la variation de l'absorption en fonction de
la longueur d'onde.
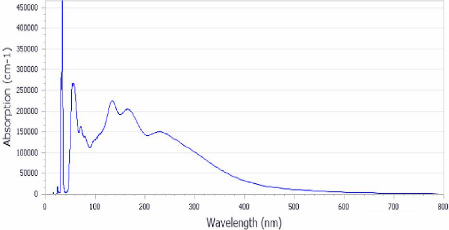
FIGURE 3.12 - L'absorbance du
TiO2 anatase calculé par DFT/GGA-PBE
Chapitre III Résultats et
discussions
Nous avons utilisé aussi le code CASTEP avec
l'approximation DFT/GGA-PBE pour décrire la fonctionnelle
d'échange-corrélation. Cette figure (3.12) nous indique des
maximums d'absorption dans le domaine ultraviolet entre 0 et 400 nm. Dans le
domaine visible entre 400 et 800 nm, l'absorption diminue jusqu'à elle
s'annule. Nous avons constaté alors, que le TiO2 anatase atteint son
maximum d'absorption juste dans le domaine ultraviolet.
La figure (3.13) montre la variation de l'intensité de
l'absorbance et de la réflectance en fonction de la longueur d'onde.
Pour la spectroscopie de transmission T et de réflexion R, nous avons
utilisé un spectrophotomètre commercial Shimadzu UV 3100 S au
design à double faisceau équipé d'une sphère
intégrée LISR 3200 capable d'enregistrer des spectres dans le
visible ainsi que dans le proche infrarouge et UV. Cette technique consiste
essentiellement en une source de lumière, un monochromateur pour
sélectionner les longueurs d'onde, un support pour conserver
l'échantillon droit par rapport au faisceau lumineux irradié et
un détecteur pour mesurer la lumière réfléchie. On
remarque que l'intensité d'absorption dans le domaine UV commence
à augmenter jusqu'à un maximum d'absorption de
7.0×101 u.a dans le domaine visible entre 400 et 800 nm. On
constate aussi un maximum d'absorption, cette absorption traduit bien la forte
aptitude d'absorber la lumière à des transitions entre les
bandes.


52 | P a g e
Figure 3.13 - Le spectre
d'absorption et de réflexion expérimentale du TiO2
anatase
Chapitre III Résultats et
discussions
53 | P a g e
La figure (3.14) montre la variation de la quantité
(??h??)2 en fonction de l'énergie
h?? pour le TiO2 obtenues
expérimentalement.

FIGURE 3.14 -
Détermination de gap optique du TiO2 anatase
Pour le calcul du gap optique on a utilisé la loi de Tauc
Plot sur la figure (3.14). En utilisant cette loi de Tauc plot sur la figure
(3.14), en traçant la tangente en un point sur l'axe des
énergies. L'intersection de la tangente avec l'axe des énergies
est la valeur de bande gap. Nous avons trouvé Eg = 3.6 eV : nous avons
trouvé aussi cette valeur précédemment avec la
méthode GGA+U : Eg = 3.601 eV.
Alors notre calcul théorique est bien en accord avec
l'expérimentale et supposé confirmé maintenant.
Le coefficient d'absorption est calculé par cette relation
:
|
On a: (??????)
|
??
?? = ??(???? - E??)??
Equation 3.4
|
Chapitre III Résultats et
discussions
54 | P a g e
Pour les transitions directes ?? = 2 , alors 1
1 ?? = 2 :
(??????)?? = ??(???? - ????)??
Equation 3.5
????????
Avec ?? = -?????? ?? et ???? =
??
??
Equation 3.6 ?? = ??.????-??????(??-??)??
??
3.5.3 Indice de réfraction :
L'indice de réfraction d'un matériau permet de
déterminer la profondeur d'absorption et par suite la profondeur des
jonctions dans les composants optoélectroniques. Cette indice mesure la
transparence des matériaux semi-conducteurs par rapport aux rayonnements
spectraux et a également un rôle important dans l'étude des
propriétés optiques. La figure (3.15) ci-dessous montre la
variation de l'indice réfraction en fonction de la fréquence.

FIGURE 3.15 - L'indice de
réfraction en fonction de l'énergie du TiO2 anatase
Chapitre III Résultats et
discussions
55 | P a g e
L'indice de réfraction peut être
déterminé à partir des parties réelle et imaginaire
de la fonction diélectrique.
Equation 3.7 ??(??) = ????(??) + ??????(??)
L'indice de réfraction réelle et le coefficient
d'extinction peuvent être donnés par les deux équations
suivantes :
??/ ? ?
v????? ?(??)+???? ??(??)
Equation 3.8 ???? (??) = [????(??)
?t + ?? ]
|
v????? ?(??)+???? ??(??)
Equation 3.9 ???? (??) = [- ????(??)
?? + ??
|
??/??
|
Le coefficient d'extinction ou l'atténuation k
représente la partie d'absorption dans l'indice de réfraction
complexe et directement lié au coefficient d'absorption.
Pour les semi-conducteurs, l'indice de réfraction
n(ù) est calculé par la partie réelle de la fonction
diélectrique.
Equation 3.10 ?? = v????
La figure (3.15) montre que le composant statique de l'indice
de réfraction n (0) = 2.63. On constate aussi que l'indice de
réfraction de la partie réelle croit de 2.63 jusqu'à 3.21
avec l'augmentation de l'énergie jusqu'à atteindre un maximum
d'énergie 2.92 eV, puis commence à diminuer.
On observe aussi presque une coïncidence entre les
positions de pics d'absorption et celles des pics du spectre de la partie
imaginaire de la fonction diélectrique.
Alors notre calcul théorique DFT/GGA-PBE est en proche
de la valeur l'expérimentale n = 2.54.
Chapitre III Résultats et
discussions
56 | Page
3.6 Résultats du la dynamique moléculaire
:
3.6.1 Détail du calcul :
La simulation de la dynamique moléculaire du dioxyde de
titane anatase a été réalisée par le code CASTEP
intégré dans Materials Studio MS. Ce choix de calcul nous a
permis d'acquérir d'avantages d'informations par rapport aux autres
modules.
Dans ce cadre, notre objectif est de prédire les
paramétres des mailles d'une part et d'une autre part pour
déterminer les structures des bandes et les densités
d'états électroniques et de les comparer avec les autres
résultats.
Comme d'habitude nous avons commencé le calcul par
l'optimisation de la géométrie. L'optimisation est
effectué par l'approximation GGA-PBE avec une énergie de coupure
450 eV.
Pour décrire les interactions d'électrons de
valence, nous avons utilisé le potentiel ultrasoft avec 3×3×3
k-points.
Nous avons obtenu la convergence après trois
itérations pour une énergie de - 9.92631981E+003 eV.
Par la suite, nous avons lancé le calcul de la
dynamique moléculaire. Nous avons utilisé l'approximation de
GGA-PBE pour décrire l'interaction d'échange -
corrélation.
Le plus important ici est que le calcul ne marche pas que si
la structure doit être dans le groupe d'espace P1 pour garantir une
meilleur relaxation structurale de tous les paramétres
cristallographiques.
Pour tenir compte des variations de la pression et de la
température, les équations du mouvement des ions et de la cellule
sont intégrées par l'algorithme de saute-mouton « Verlet
Leap-Frog »
La configuration initiale correspond à la
température minimale, choisie pour être identique à celle
de la structure expérimentale. Le systéme est alors
équilibré à la pression et la température de
l'atmosphère (T= 300K).
Nous avons adopté aussi l'algorithme de Nosé -
Anderson - Barostat de l'ensemble thermodynamique NPT.
Notre systéme est intégré de 100 pas
d'intégration avec un pas Lt = 1 ???? .
La figure (3.16) présente la structure anatase
optimisé au cours de calcul de la dynamique moléculaire.

Chapitre III Résultats et
discussions
57 | Page
FIGURE 3.16 - La structure
anatase en cours de calcul dynamique
Au cours de calcul, cette structure de la figure (3.16), nous
avons marqué des fluctuations et des vibrations de cette structure. Ce
calcul a duré plusieurs heures il y a environ 9 heures. Nous avons
contrôlé notre structure et nous avons constaté qu'elle
commence à déformer légèrement. La figure (3.17)
ci-dessous, montre la variation de la constante du mouvement en fonction du
temps de la simulation. On remarque que cette constante décroit
progressivement de -9756.94 jusqu'à -9756.99, puis une
légère augmentation et enfin un chute brisque de -9756.96
jusqu'à elle s'annule.

FIGURE 3.17 - Evolution de la
constante du mouvement en fonction du temps de simulation
Chapitre III Résultats et
discussions
La figure suivante (3.18) montre l'évolution de la
pression au cours de la simulation en fonction du temps de calcul. Cette figure
indique une augmentation continue de la pression jusqu'à elle atteint 36
GPa.

FIGURE 3.18 - Evolution de la
pression en fonction du temps de simulation
La figure (3.19) montre aussi l'évolution de la
température en fonction du temps de la simulation.

FIGURE 3.19 - Evolution de la
température en fonction du temps de simulation
58 | P a g e
Chapitre III Résultats et
discussions

200 GPa
59 | P a g e
La température varie entre 0 et 450 K et le temps de la
simulation varie entre 0 et 0.1 pc. Pendant ces intervalles la
température initiale est 300 K comme que déjà la fixer,
puis elle commence à varier toute l'intervalle. On constate que la
température al fin de la simulation est presque celle de la
température initiale 300 K, cela nous indique que le systéme a
subit la relaxation à la fin de cycle et donc un retour à
l'état d'équilibre de systéme.
3.6.2 Paramétres de mailles :
La figure (3.20) ci-dessous montre une partie du statut de
calcul obtenue à la fin de la simulation. Cette fichier est très
importante, on trouve les informations de calcul les tous
détaillées. Cette partie est la partie spécifique pour les
paramètres des mailles à prédire par la dynamique
moléculaire. Nous avons lancé deux calculs l'un avec 200 GPa et
l'autre 100 GPa.

100 GPa
FIGURE 3.20 - Statut de calcul
des paramétres de mailles
Chapitre III Résultats et
discussions
60 | P a g e
Ces valeurs trouves sont en accord avec les paramétres
des mailles trouvés expérimentalement. Sur le tableau suivant
(3.6), nous avons présenté tous les paramétres des mailles
de notre calcul et les valeurs trouvées par l'expérimentale.
|
Paramètres des
mailles
(A°)
|
Expérimentale
|
Notre calcul Notre calcul
Avec 200 GPa Avec 100 GPa
|
|
a
|
3.782
|
3.639115 3.632904
|
|
b
|
3.782
|
3.639115 3.632904
|
|
c
|
9.502
|
9.142120 9.126518
|
|
á
|
90
|
90 90
|
|
fi
|
90
|
90 90
|
|
ã
|
90
|
90 90
|
TABLEAU 3.6 - Les
paramétres des mailles calculés et expérimentales
Le tableau (3.6) nous montre aussi que si on a diminué
la pression de 200 à 100 GPa, les paramétres des mailles a, b et
c diminuent légèrement.
En contraire, les angles des mailles restent inchangés
pour les deux valeurs de pression. Alors on peut constater que la pression est
un facteur très important qui influe directement sur les
paramètres des mailles. Donc la pression peut varier ces
paramétres uniquement pour a, b et c et pas pour les angles.
3.6.3 Structure des bandes et densité
d'état électronique DOS :
Les figures ci-dessous (3.21) et (3.22) montrent
respectivement les structures des bandes pour 200 GPa et pour 100 GPa et les
densités d'états électroniques DOS.
Le calcul de ces propriétés est
réalisé par le code CASTEP inclue dans MS avec la même
énergie de coupure 450 eV.
L'interaction d'échange - corrélation est
décrite par l'approximation de GGA - PBE avec un K-Points
3×3×3.
Chapitre III Résultats et
discussions

200 GPa
100 GPa
FIGURE 3.21 - Les structures des
bandes pour deux pressions
61 | P a g e
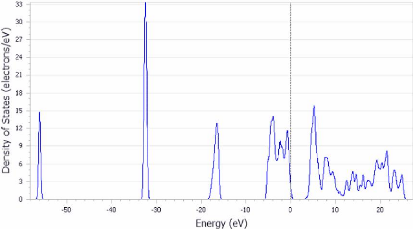
Chapitre III Résultats et
discussions
62 | P a g e
200 GPa

100 GPa
FIGURE 3.22 - Les densités
d'états électroniques DOS
La première figure montre un gap de 3.701 eV pour une
pression de 200 GPa et la deuxième
montre un gap de 3.843 eV pour la
pression de 100 GPa. Ceux valeurs trouves sont un peu loin
Chapitre III Résultats et
discussions
63 | P a g e
de gap expérimentale obtenue 3.6 eV et aussi peu loin
des valeurs calculés par DFT. Mais on constate bien que si on a
diminué la pression de 100 à 200 GPa, le gap à augmenter
automatiquement.
Alors on peut comprendre que la pression est un facteur
très important influant directement sur la largeur des bandes
interdites. Donc cela nous permet de passer d'un semi-conducteur à un
isolant ou d'un isolant a un semi-conducteur.
Conclusion générale
65 | P a g e
Dans ce travail, nous avons
étudié les propriétés électroniques et
optiques de dioxyde de titane anatase pur. Nous avons utilisé la
méthode des ondes planes et du pseudo-potentiel dans le cadre de la DFT
avec les approximations GGA-PBE, GGA+U, GGA-PBEsol et B3LYP, en utilisant le
code CASTEP implémenté dans le logiciel Materials Studio.
D'une part, ensemble des calculs
théoriques de la DFT obtenus sont en accord avec d'autres calculs
théoriques basés sur la théorie de DFT. Les calculs de
structures de bandes électroniques pour le dioxyde de titane anatase
révèlent la présence d'un gap énergétique
direct. Il faut remarquer que la valeur de gap énergétique
déterminée à partir de l'approximation GGA-PBE est faible,
ceci est une limitation bien connue de la (DFT).
Cependant l'utilisation de l'approximation
GGA+U a amélioré les valeurs des gaps de façon
considérable, ce qui nous rapproche des résultats
expérimentaux.
Les résultats obtenues de spectre Raman montrent que la
fonctionnel hybride (B3LYP) est très précise que les autres
approximations.
D'une autre part, nous avons calculé
aussi les paramétres des mailles, les structures des bandes et les
densités d'états par la simulation de la dynamique
moléculaire. Nous avons trouvé des résultats très
comparables et acceptables.
Dernièrement, les fonctionnels
hybrides sont les plus performants et les plus exactes pour la simulation. Mais
aussi il existe d'autres générations des fonctionnelles dite
fonctionnelles métahybrides qui sont encore plus puissant.
66 | P a g e
Bibliographie
[1] A. Russell, «The
mineralogical magazine» The Mineralogical Magazine and Journal of the
Mineralogical Society, n° %1229, pp. 617-624, 1955.
[2] S. V. S. V. R. J. M. Subramanian
«Effect of cobalt doping on the structural and optical
properties of TiO2 films prepared by sol-gel process» Thin Solid Films,
vol. 516, pp. 3776-3782, 2008.
[3] D. A. H. H. a. C. C. Sorrell
«Review of the anatase to rutile phase
transformation,» Journal of Materials Science, vol. 46, n° %14, p.
855-874, Dec 2010.
[4] H. PERRON «
Thése de Doctorat, " Simulation par la théorie de la
fonctionnelle de la densité de l'interaction de l'ion uranyle avec les
surfaces de TiO2 et de NiFe2 O4,» Université Paris sud, Orsay,
2007.
[5] C.-H. H. H.-W. C. Yi-Mu
Lee, « Structural, optical, and electrical properties of
p-type NiO films and composite TiO2/NiO electrodes for solid-state
dye-sensitized solar cells» Applied Surface Science, vol. 255, pp.
4658-4663, 2009.
[6] B. M. Ihsèn,
«Elaboration et caractérisation physique des couches minces de TiO2
déposées par pulvérisation cathodique» Diplôme
de Mastère, Tunis, 2009.
[7] †. C. T. D. L. H. H. a. T. I. Ji-Guang
Li, «Monodispersed Spherical Particles of Brookite-Type
TiO2 : Synthesis, Characterization, and Photocatalytic Property» Journal
of the American Ceramic Society, n° %187, pp. 1358 -1361 , 2004.
[8] S. B. e. E. Houdeau,
«Exposition orale aux nanoparticules de dioxyde» Journale de Biologie
Aujourdhui, vol. 208, n° %12, pp. 167-175, 2014.
[9] P. H. a. W. Kohn,
«Inhomogeneous electron gas» Journal of Phys Rev, vol. 136, p. B864-
B871, Nov 1964.
[10]
67 | P a g e
2. P. J. D. L. 7. M. J. P. C. J. P. P. J. H. S. J. C.
a. M. C. P. M D Segall1, «First-principles simulation :
ideas, illustrations and the CASTEP code» Journal of physics : Condensed
Matter, p. 2717-2744, 2002.
[11] M. B. a. R. Oppenheimer,
«On the Quantum Theory of Molecules», Journal of Ann. Phys, n°
%184, p. 485, 1927.
[12] P. H. a. W. Kohn,
«Inhomogeneous electron gas», Journal of Physical Review, vol. 136,
n° %13 B, Nov 1964.
[13] K. Korchagina, «Etude
par dynamique moléculaire des propriétés structurales,
dynamiques et thermodynamiques d'agrégats moléculaires»,
Université Paul Sabatier, Toulouse, 2016.
[14] 2. P. J. D. L. 7. M. J. P. C. J. P. P. J. H.
S. J. C. a. M. C. P. M D Segall1, «First-principles
simulation : ideas, illustrations and the CASTEP code», Journal of physics
: Condensed Matter, p. 2717-2744., 2002.
[15] T. DAS, «Theoretical
study of the electronic and optical properties of photocatalytic inorganic
materials,» Université de Nantes UFR sciences et techniques,
Nantes, 2012.
[16] W. Y. R. G. Parr,
«Density Functional Theory of Atoms and Molecules», International
Journal of Quantum Chemistry, vol. 47, n° %1101, p. 333, 1989.
[17] V. BADAUT, «Approche
couplée chimique, spectroscopie et de modélisation ab initio
à la réactivité de surface application à la
rétention des anions par la sidérite,» Université
Paris-Sud XI U.F.R Scientifique d'Orsay, Orsay, 2010.
[18] Z.-Y. Z. Yong-Dong Zhou,
«Interfacial structure and properties of TiO2 phase junction studied by
DFT calculations,» Journal of Applied Surface Science, vol. 485, p. 8 - 21
, 2019.
[19] J. D. Meili Guo,
«Principles study of electronic structures and optical properties of Cu,
Ag, and Au-doped anataseTiO2» Physica B : Condensed Matter, vol. 407, pp.
1003-1007, 2011.
[20]
68 | P a g e
J. F. D. C. F. C. M. J. F. P. J.
Stephens, «Ab Initio Calculation of Vibrational
Absorption and Circular Dichroism Spectra Using Density Functional Force
Fields,» The Journal of Physical Chemistry , vol. 98, n° %195, pp.
11623-11627, 1994.
[21] H. PERRON,
«Simulation par la théorie de la fonctionnelle de la densité
de l'interaction de l'ion uranyle avec les surfaces de TiO2 et de NiFe2
O4,» Université Paris Sud - Paris
X, Paris, 2007.
[22] I. V. M.G. Brik n,
«First-principles calculations of optical and electronic properties of
pure andSm3+-doped TiO2,» Journal of Physica B, vol. 405, p. 2450-2456,
2010.
[23] I. M. D. S. C. J. P. P. J. H. M. I. J. P. K.
R. M. C. P. Stewart J. Clark, «First principles methods
using CASTEP,» Journal of Z. Kristallogr, vol. 220, pp. 567-570,
2005.
[24] J. J. Y. S. Toshihiro
Okajima, «Geometric structure of Sn dopants in sputtered
TiO2 film revealed by x-ray absorption spectroscopy and first-principles DFT
calculations,» Journal of Materials Research Express, vol. 5, n° %14,
2018.
[25] P. Z. J. L. a. J. Y. Jinfeng
Zhang, «New understanding of the difference of
photocatalytic activity among anatase, rutile and brookite TiO2,» Journal
of Physical Chemistry Chemical Physics, vol. 16, pp. 20382-20386, 2014.
[26] Q. L. Zongyan Zhao,
«Effects of lanthanide doping on electronic structures and optical
properties of anatase TiO2 from density functional theory calculations,»
Journal of Physics D : Applied Physics, vol. 41, p. 10, 2008.
[27] Y. D. , B. H. Kesong Yang,
«First-principles calculations for geometrical structures and electronic
properties of Si-doped TiO2,» Journal of Chemical Physics Letters, vol.
456, p. 71-75, 2008.
[28] A. D.H, «The relation
between the Raman spectra and the structure of organic molecules,» Journal
of Physics Review, vol. 36, pp. 544-554, 1930.
[29] M. D. S. C. J. P. P. J. H. M. I. J. P. K. R.
M. C. P. Stewart J. Clark, «First principles methods
using CASTEP,» Z. Kristallogr, vol. 220, p. 567-570., 2005.
[30]
69 | P a g e
T. T. Thao, «Thése :
Etude par dynamique moléculaire de spectres vibrationnels de verres de
silice,» Université des Sciences et Technologie de Lille - Lille I,
Lille, 2007.
[31] L. Verlet, «Computer
"Experiments" on Classical Fluids. I. Thermodynamical Properties of
Lennard-Jones Molecules,» Journal of Physical Review, vol. 159, n°
%11,
p. 98, Juillet 1967.
[32] D. Poger,
«Thése : Structure dynamique moléculaire et
sélectivité de métallochaperones à cuivre et
à mercure,» Université Joseph Fourier, Grenoble,
2005.
[33] T. L. T. NGUYEN,
«Thése : Etude par dynamique moléculaire de l'alliage
eutectique Au-Si en volume et en interaction avec un substrat de
silicium,» Université de Grenoble,, Grenoble, 2012.
[34] Q. M. H. Luo, «Molecular
Dynamics Simulation of Sintering Dynamics of Many TiO2 Nanoparticles,»
Journal of Statistical Physics, vol. 160, n° %16, pp. 1696-1708,
2015.
[35] P. A. F. a. T. C. D. ] S. P. S.
Porto, «Raman spectra of TiO2, MgF2, ZnF2, FeF2, and
MnF2,» Physical Review , vol. 522, p. 154, 1967.
[36] T. J.-n. GONG Xiao-zhong,
«Preparation and characterization of La-Co alloy nanowire arrays by
electrodeposition in AAO template under nonaqueous system,»
Trans.Nonferrous Met.Soc.China, pp. 642-647, 2008.
[37] U. B. a. N. G. Eror,
«Raman Spectra of Titanium Dioxide,» solid state chemistry , vol. 42,
pp. 276-282, 1982.
[38] B. V. a. U. V. V. T
Maiyalagan, «Fabrication and characterization of uniform
TiO2 nanotube arrays by sol-gel template method,» Bull. Mater. Sci, vol.
2.
[39] E. Vasseur,
«Thése Mise au point d'une procédure de dosage de TiO2 par
spectroscopie Raman,Etude de processus de rutilisation dans un four
industriel», l'université des sciences et technologie de Lille,
Lille.
[40] L. B.-G. Gong Sai,
«Electronic structures and optical properties of TiO2 : Improved
density-functional-theory investigation,» Chin. Phys. B, vol. 21, n°
%5, 2012.
| 

