V. Matériel et méthode
V.1 Analyse des terres et des argiles
Afin de répondre aux objectifs fixés, les
échantillons vont être caractérisés d'un point de
vue chimique, minéralogique et physique. La caractérisation
chimique consistera à déterminer la composition chimique
élémentaire par fluorescence X. La caractérisation
minéralogique se fera par diffraction des rayons X. La
caractérisation physique consistera à déterminer la teneur
en eau naturelle, la teneur en matières organiques, la masse volumique,
les limites d'Atterberg, et la granulométrie.
Diffractométrie des rayons X
(DRX)
Préparation des échantillons
Une analyse sur poudre totale et sur la fraction argileuse a
été réalisée sur chaque échantillon. La
préparation des échantillons s'est faite selon le protocole du
laboratoire Argiles, Géochimie et Environnements sédimentaires de
l'Université de Liège (Fagel, 2010) adapté selon Moore et
Reynolds (1989).
Pour la poudre totale, les échantillons ont
été séchés à l'étuve à
30°C, broyés manuellement dans un bol en agate, puis tamisés
pour récupérer les fractions inférieures à 250
ìm. On a ensuite constitué des pastilles à l'aide des
supports métalliques pour la diffractométrie. Pour la
minéralogie de la fraction argileuse, nous avons réalisé
un agrégat orienté. Un agrégat orienté est un
dépôt de particules argileuses inférieures à 2
ìm sur une lame de verre (Moore et Reynolds, 1989). L'analyse par
Diffraction des Rayons X (DRX) des minéraux argileux étant
basée sur la connaissance des distances réticulaires (001), on
cherche à renforcer les réflexions (001) en orientant les
particules (Moore et Reynolds, 1989). Pour se faire, une petite portion de
chaque échantillon séché est placée dans un berlin
et mélangée à l'eau distillée, puis
homogénéisée par agitation sur une plaque
magnétique. Une fois l'échantillon bien
homogénéisé, s'en est suivi un tamisage à 63
ìm, en présence d'eau.
Le tamisat est récupéré et laissé
au repos pendant 50 minutes pour que la sédimentation s'effectue. On
récupère enfin à l'aide d'une pipette de 1,5 cc à
la profondeur de 1 cm la fraction inférieure à 2 ìm. Cette
portion est posée sur une lame de verre de 25 x 25 mm et laissée
sécher pendant au moins 12 heures.Les lames orientées obtenues
pour chaque échantillon sont analysées au diffractomètre
pour fournir les diffractogrammes dits diffractogrammes à l'état
normal (N).
Certaines argiles ont des distances réticulaires
voisines, et sont donc difficiles à différencier sur un
diffractogramme orienté à l'état normal. C'est pourquoi,
toutes les lames orientées à l'état normal ont subi deux
nouveaux traitements :
- la solvatation à la vapeur
d'éthylène-glycol (EG) : se fait sous cloche et a pour
conséquence de faire gonfler les feuillets de certains minéraux
argileux (Fagel, 2010) ;
- le chauffage à 500°C : permet de
déshydrater le minéral argileux (Fagel, 2010).
Ces deux traitements modifient l'espace interfoliaire de
manière spécifique et permettent de différentier les
espèces argileuses entre elles (Fagel, 2010).
Méthode d'analyse
L'échantillon de poudre placé sur lame de verre
est bombardé par des rayons X obtenus en bombardant une anode de cuivre
par un faisceau d'électrons accélérés dans le vide.
Ces rayons X au contact avec la poudre sont réfléchis par chaque
cristallite de la poudre selon une orientation dans l'espace. Les rayons X
réfléchis interfèrent entre eux avec diverses
intensités (Tucker et Hardy, 1988). On enregistre l'intensité
détectée en fonction de l'angle de déviation 2è
(deux-thêta) du faisceau. La courbe obtenue s'appelle diffractogramme ou
spectre DRX. En analysant les spectres DRX obtenus, il est possible de
déterminer les phases minérales qui constituent la poudre. En
effet, pour certains angles de déviation 2è du faisceau, on
détecte des rayons X. Ce sont les « pics » du diffractogramme.
Ces angles de déviation sont caractéristiques de l'organisation
des atomes dans la maille cristalline.
35
Dans les autres directions, on ne détecte pas de rayon
X, c'est la ligne de fond du signal (Tucker et Hardy, 1988). Si l'on calcule
les directions dans lesquelles on a du signal, on s'aperçoit que l'on
obtient une loi très simple :
2.d.sin(è) = n.ë
où è est la moitié de l'angle de
déviation, n est un nombre entier appelé «ordre de
diffraction», ë est la longueur d'onde des rayons X et d la distance
entre les plans d'alignement des atomes ou distance interréticulaire.
C'est la loi de Bragg (Tucker et Hardy, 1988).
Par la suite, le logiciel EVA a été
utilisé pour lire les diffractogrammes et permettre d'identifier les
phases minérales, grâce au paramètre d.
Analyse chimique par Fluorescence X
(FX)
La spectrométrie de fluorescence X (SFX ou FX) est une
technique d'analyse chimique élémentaire d'un échantillon.
Elle constitue une des premières étapes dans la
caractérisation de tout matériau et permet d'éclairer les
propriétés des échantillons argileux en vue d'une
utilisation dans diverses applications.
Principe
Lorsque l'on bombarde de la matière avec des rayons X,
celle-ci réémet de l'énergie sous la forme de rayons X,
dont le spectre est caractéristique de la composition de
l'échantillon. En analysant ce spectre, on peut en déduire la
composition élémentaire (les concentrations massiques en
éléments).
Appareillage
Le spectromètre utilisé est de marque ARLTM
PERFORM'X du laboratoire de pétrologie sédimentaire (PETROSED) de
l'ULg.
Préparation des échantillons
Le matériau est séché à 40°C,
broyé et tamisé à 250 um. La perte de masse par chauffage
est d'abord déterminée. Pour cela, l'échantillon est
chauffé à une température de 1000 °C pendant 2
heures. Cette température est atteinte en 4 heures. Il est ensuite
décomposé par un broyage manuel, puis mélangé
à une proportion de 11x sa masse (+/- 0,0003) au flux de borax
(Na2B4O7.10H2O) et à environ 0,002 gr de LiBr. Ces composés ont
pour propriété de baisser la température de fusion. Les 3
composants (borax, LiBr et échantillon) sont ensuite
mélangé manuellement puis soumis à la fusion en s'assurent
une bonne homogénéité. La dernière étape
consiste à réaliser des perles qui seront analysées.
Étant donné qu'un solvant de borax a été
utilisé, l'oxyde de sodium Na2O n'est pas dosé. Les
concentrations élémentaires ont été obtenues en
réalisant une calibration par rapport à des standards (Adler,
1966 ; Lachnitt, 1983; Müller, 1972).
Analyse granulométrique et
sédimentométrie
L'analyse granulométrique permet de classer les grains
d'un échantillon selon leurs tailles, et donne le pourcentage de chaque
classe par rapport au poids total de l'échantillon. L'échantillon
à classer est passé dans une série de tamis emboîtes
ayant des diamètres d'ouverture croissants du bas vers le haut : 2,38mm
et 4,76 mm. L'échantillon est placé dans le tamis
supérieur et le classement se fait par vibration manuelle ou à
l'aide d'une plaque vibrante.
Le refus récupéré dans chaque tamis est
alors pesé et le pourcentage déterminé. Le résultat
est reporté sur une courbe granulométrique.
La sédimentometrie est la technique la plus
adaptée pour repartir en classes granulométriques les
éléments plus fins que 75 ìm. L'échantillon est
mélangé à un défloculant (le sulfate de sodium)
afin de séparer les colloïdes ; et à l'eau. Le tout est
mixé puis place à 20°C pendant 24h avant d'être
homogénéise par agitation manuelle.
La vitesse de sédimentation des particules solides (v)
est liée à leur diamètre (D), à leur masse
spécifique (Ys), à la masse spécifique du liquide qui les
contient (Yl) et à la viscosité de ce même liquide
(ç), selon la relation :
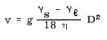
36
Au temps t après le début de la
sédimentation, la suspension ne contient à la profondeur h de la
surface, que des particules d'une certaine vitesse et d'un certain
diamètre. Le pourcentage en poids de ces particules à cet instant
peut être calculé au moyen d'un aéromètre en tenant
compte des corrections liées à la température ou encore au
défloculant (Peltier et Rumpler, 1959).
Limites de consistance
Les limites de consistance ou limites d'Atterberg
correspondent aux proportions en eau pour lesquelles le matériau
argileux passe d'un comportement semi-liquide à un comportement
plastique (limite de liquidité, WL), et ensuite d'un comportement
plastique à un comportement semi-solide (limite de plasticité WP)
(fig. II.3). Les limites d'Atterberg servent à classer les sols, et
à prévoir leur comportement lorsqu'ils sont sollicités
mécaniquement (Peltier et Rumpler, 1959).
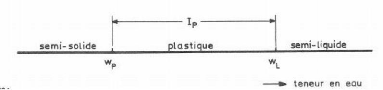
Fig. II.3 : Limites de
consistance (Centre de recherches routières, 1981).
Limite de liquidité
On prend 200 gr de matériaux de granulométrie
inférieure à 425 ìm, on y ajoute de l'eau pour bien
homogénéiser et former une pâte argileuse. Celle-ci est
placée ensuite dans une coupelle et on définit une lèvre
calibrée. Ensuite l'échantillon est secoué sur l'appareil
de Casagrande, et on note le nombre de coups correspondant à la
fermeture de la lèvre sur 1 cm. On s'efforce à avoir toujours un
nombre de coups compris entre 15 et 35. Les mesures sont reportées sur
un graphique teneur en eau - nombre de coups.
Limite de plasticité
On utilise la pâte argileuse pour former un petit boudin
de 3 ou 4 cm de long, puis on le roule sur une surface plane jusqu'à ce
qu'il atteigne une longueur d'environ 10 cm de long et 3 mm de diamètre
à sa rupture. La limite de plasticité correspond à la
teneur en eau du boudin à sa rupture.
On répète la manipulation 3 fois et la limite de
plasticité correspond à la valeur moyenne de ces 3 mesures.
Indice de plasticité
Elle caractérise l'intervalle où le
matériau argileux demeure plastique ou façonnable. Elle s'obtient
par la différence entre la limite de liquidité et de
plasticité : Ip = WL - WP
Masse volumique
La mesure de la masse volumique s'est faite à l'aide
d'un pycnomètre à gaz suivant la norme NF EN ISO 8130-2 au
laboratoire de matériaux de construction de l'ULg. Ce pycnomètre
est un moyen rapide de mesurer avec précision la masse volumique et la
porosité d'un matériau. Cet appareil utilise un gaz
(l'hélium dans notre cas) à une pression variant entre 140-170
kPa. L'échantillon à analyser, de masse connue Me, est introduit
dans la petite cellule en aluminium du pycnomètre. L'hélium est
alors confiné à une certaine pression P1 dans le volume V1 connu
d'une cellule. Le gaz est ensuite libéré dans le volume de
détente V2 dans lequel se situe l'échantillon. On obtient alors
une certaine pression P2. Le volume Ve de l'échantillon peut être
déterminé grâce à la loi des gaz parfaits. La mesure
est
37
effectuée quatre fois et le résultat final
correspond à la valeur moyenne.
Perte au feu
La perte au feu (LOI, Loss On Ignition) est la perte de poids
de l'échantillon par processus de déshydratation
(élimination de l'eau de constitution des minéraux,
élimination de l'humidité résiduelle,...) et la
disparition de la matière organique.
Principe
L'échantillon est chauffé dans un four pendant
24 heures à 105 °C afin de le déshydrater. La
différence des pesées entre l'échantillon non
chauffé et chauffé donne la teneur en eau. Ensuite, la
matière organique est éliminée sous formes de cendres et
de CO2 durant une chauffe de 4 heures à 550 °C dans un second four
(mesures réalisées au laboratoire AGEs de l'ULg). L'analyse dans
notre cas s'est faite sur un échantillon de terre, d'environ 1,5 gr que
l'on a placé dans un bol en céramique d'environ 10 ml.
Exploitation des données
La teneur en eau est calculée selon :
)*100
La densité sèche est déduite de la teneur en
eau :
densité sèche = (100 - teneur en eau)/100 x
densité du grain
Cette formule considère que l'échantillon contient
de l'eau et des particules sédimentaires.
La densité est fonction du type de grain en
g/cm3 : Calcite 2,71 ; Quartz 2,65 ; Argiles 1,8-2,2 ; Opale 1,4 ;
Eau 1 ; ...
La teneur en matière organique est calculée selon
la perte au feu à 550 °C suivant :
Le carbone organique total peut être estimé à
50 % de la MO totale (Tucker et Hardy, 1988). V.2
Caractérisation des liants
Les liants sont des minéraux souvent classifiés
en deux catégories : les liants hydrauliques (ciment Portland, chaux
hydraulique, laitiers, ciments spéciaux, etc.) et les liants
aériens (chaux aérienne, gypse, plâtre, argile, etc.), qui
diffèrent selon la manière dont ils durcissent. Les liants
hydrauliques durcissent grâce à la réaction qu'ils
développent avec l'eau alors que les liants aériens ont besoin de
CO2 pour durcir par carbonatation (Van Balen, 2005).
La chaux
Parmi la chaux, on peut spécifier deux types : la chaux
hydraulique et la chaux aérienne, qui diffèrent selon le
processus de production, et donc du matériau brut utilisé. La
chaux aérienne est obtenue par la calcination de calcaire naturel ou de
coquillages alors que la chaux hydraulique est produite à partir de
calcaire impur, contenant des composés argileux ou organiques ou des
mélanges de calcaire et d'argile. Dans cette étude, seule la
chaux hydraulique sera traitée dans la phase expérimentale.
Les pouzzolanes
Les pouzzolanes sont des matériaux n'ayant aucune
capacité propre de liant mais pouvant réagir avec de la chaux
Ca(OH)2 en présence d'eau à température ambiante afin de
former des composants du
38
ciment. Ce pouvoir est appelé « activité
pouzzolanique ».
On distingue :
- les pouzzolanes naturelles : proviennent
généralement de l'activité volcanique mais aussi de la
sédimentation consolidée. C'est par exemple la terre de Santorin,
le tuff et le trass de la région du Rhin en Allemagne, la
Bavière, la Roumanie et la pierre ponce de l'Eifel en Allemagne.
(Verhasselt, 1993)
- les pouzzolanes artificielles obtenues par calcination de
certaines argiles, schistes argileux et autres diatomites. Ils possèdent
les mêmes propriétés que les pouzzolanes naturelles
(Verhasselt, 1993).
Dans le cas des liants chaux/pouzzolane, les pouzzolanes
réagissent avec le Ca(OH)2 de la chaux hydratée. La vitesse
globale de la réaction pouzzolanique dans les liants dépend de
nombreux facteurs (Hewlett, 2004) : la quantité de pouzzolane dans le
liant ; la quantité de SiO2 dans la pouzzolane ; la nature des phases
actives dans la pouzzolane ; la surface spécifique de la pouzzolane ;
les propriétés physiques et chimiques du ciment et/ou de la chaux
hydratée ; la durée de durcissement ; la température ; le
rapport eau/solide.
La cendre de balle de riz
Composition chimique
D'une manière générale, la teneur en
silice augmente lorsque la température et/ou la durée de
combustion augmente, et lorsque la perte au feu, liée à la
quantité de carbone, diminue (Sabuni, 1995). Les impuretés
principales de la cendre sont des produits alcalins dont le potassium est le
constituant prédominant (Zhang et al., 1996). Un changement de couleur
est observé en fonction de la teneur en carbone (allant du noir vers le
blanc, voir le rose pâle lorsqu'il n'y pas plus la présence de
carbone. Composition minéralologique
Pour obtenir une cendre pouzzolanique, la CBR doit contenir de
la silice amorphe (Feng et al., 2004). Durant la calcination, la cendre peut se
cristalliser donnant des faibles propriétés pouzzolaniques.
Toutes les recherches ont montré que pour l'éviter, la
température de combustion ne doit pas excéder 750°C. Halleux
(2012), a effectué une représentation qualitative des phases
amorphes et cristallines de la silice par DRX sur trois échantillons de
cendres de balles de riz calcinées à des températures
différentes : 600, 700 et 800°C. Le spectre a montré des
raies correspondant aux distances inter-réticulaires du quartz (3,34 ;
4,26 ; 1,82 et 1,54), de la cristobalite (4,04 ; 2,49 et 2,47) et de la
tridymite (4,11 et 4,33). Il a remarqué que :
- la cendre obtenue à 600°C présentait une
distance inter-réticulaire « d » proche de 4,20311 pouvant
expliquer la présence de cristobalite et dont l'intensité
était plus faible que pour celui représentant la cristobalite
à 700°C. Cela explique donc la formation de phases cristallines
lorsque la température augmente.
- entre la DRX caractérisant la cendre calcinée
à 600°C et la DRX pour une cendre calcinée à
800°C, la quantité de quartz diminue. Il y a donc transformation du
quartz en phase cristalline.
Il a donc choisi la température de 600°C pour
calciner la balle de riz.
Calcination
Durant la combustion, la matière organique est
brûlée, produisant une cendre de balle dont la masse est d'environ
20% de la masse initiale des balles. Il s'agit d'un matériau poreux,
riche en silice mais dont les propriétés dépendant
beaucoup des conditions de calcination (Feng et al., 2004). Ainsi, la cendre
produite par des incinérateurs à hautes performances en
laboratoire peut généralement atteindre de haut pourcentage de
silice pouvant atteindre 98 % (Halleux, 2012). La calcination s'est faite
suivant un protocole proposé par Halleux (2012). Elle utilise un four
pouvant atteindre de hautes températures. Une quantité de 2,2 kg
est calcinée à chaque fournée de 2 heures. On obtient
ainsi à chaque fois 400 g de cendres environ. Lors de la calcination,
une couche de carbonisation se forme tout autour du profilé, ce qui
empêche la balle de riz située à la
périphérie de prendre correctement feu. Pour palier à
cela, un mélange fréquent est recommandé afin de casser
cette couche de
39
carbonisation. Un mélange a été
réalisé toutes les 30 minutes pour obtenir une cendre de couleur
quasi homogène. La température de calcination est de
600°C.
Surface spécifique
La surface spécifique de la CBR dépend aussi des
conditions de calcination (température, durée). En effet, la
cristallisation de la silice mène à l'agglomération des
particules, alors que la présence de carbone dans la cendre, augmente la
surface spécifique étant donné que le carbone est
très poreux (Jaturapitakkul et Roongreung, 2003). La surface
spécifique peut être contrôlée par le broyage. En
broyant la CBR, on diminue la taille des particules et on augmente la surface
spécifique. La structure poreuse se casse et donne lieu à des
fines particules poreuses possédant des propriétés
similaires de celles de la fumée de silice (Sabuni, 1995). Mais le
broyage ne doit pas être prolongé afin d'éviter la
destruction complète de la structure poreuse et une agglomération
des particules diminuant ainsi la surface spécifique de la CBR (Bui et
al., 2005). La surface spécifique de la CBR influence la réaction
pouzzolanique avec de la chaux. Elle est plus importante lorsque la surface
spécifique augmente (Feng et al., 2004). La grande surface
spécifique demandera également une quantité importante
d'eau pour obtenir une bonne mise en oeuvre du mélange incorporant la
balle de riz (Feng et al., 2004).
La granulométrie de la CBR a été
réalisée après un broyage et un tamisage manuels.
Dans notre étude, nous avons choisi les tamis à
250ìm, 150ìm, 75ìm et 53ìm. Cela nous a donc permis
d'obtenir une cendre avec une granulométrie proche des valeurs
trouvées dans la littérature : < 250 um : 95 % ; < 150 um :
85 % ; < 75 um 67 % ; < 53 um : 54 %.
Activité pouzzolanique
Feng et al., 2004 ont indiqué que la réaction
pouzzolanique augmentait avec l'accroissement de la quantité de silice
amorphe et l'augmentation de la surface spécifique. Différentes
méthodes existent pour déterminer l'activité
pouzzolanique. Beaucoup reposent sur le concept de la variation de
conductivité électrique, et montrent que la cendre de balles de
riz a une activité pouzzolanique similaire à celles obtenues pour
d'autres pouzzolanes artificielles (cendres volantes et fumées de
silice). (Bui, 2001 ; Feng et al., 2004).
L'activité pouzzolanique de la CBR a été
effectuée par la méthode de Luxan et al. (1989). C'est une
méthode indirecte et qualitative basée sur la mesure du
changement de conductivité électrique. Luxan et al. (1989)
imposent une solution saturée en chaux Ca(OH)2 de 200 ml à une
température de 40°C avec 5g de cendres. On mesure tout d'abord la
conductivité de la chaux seule dans de l'eau
déminéralisée, via les ions Ca2+ et les ions
OH- en provenance du Ca(OH)2 . Ensuite on ajoute la CBR. La silice
amorphe commence à réagir avec les Ca(OH)2 et forme des
C-S-H1. Cette réaction réduit le nombre d'ions de
Ca2+ et OH- présents dans la solution et diminue
la conductivité électrique. La variation de la
conductivité électronique est calculée en soustrayant la
valeur de la conductivité de la solution eau-chaux par la somme des
conductivités des solutions eau-chaux-CBR et eau-CBR. D'après
Luxan et al. (1989), un matériau est considéré comme ayant
une mauvaise activité pouzzolanique lorsque la variation de
conductivité est inférieure à 0,4 mS/cm ; une
pouzzolanicité variable lorsque la variation se situe entre 0,4 et 1,2
mS/cm ; et une bonne pouzzolanicité lorsque la variation est
supérieure à 1,2 mS/cm.
La CBR a été testée deux fois, et la valeur
moyenne a été sélectionnée (tableau V.1).
1 C-S-H (Calcium silicate hydrate) est le résultat de la
réaction entre la chaux, la silice et l'eau 3Ca(OH)2 + 2SiO2 + H2O ?
3CaO . 2SiO2 . 3H2O + H2O
40
Conductivité solution
chaux [mS/cm]
|
Conductivité chaux +
cendres [mS/cm]
|
Conductivité eau +
cendres [mS/cm]
|
Variation de
conductivité électrique
|
|
7,89
|
5,25
|
0,759
|
1,896
|
|
7,88
|
5,33
|
0,675
|
1,859
|
|
Moyenne 1,878
|
Tableau V.1 : Activité
pouzzolanique obtenue par la méthode de Luxan et al. (1989).
Liants chaux - CBR
La CBR pouzzolanique réagit avec le Ca(OH)2
présent dans la chaux hydratée. Cela entraîne la formation
de produits stables et insolubles dont principalement des phases de C-S-H. La
quantité de Ca(OH)2 ou d'ions Ca2+ est donc moindre quand il
y a une addition de CBR (Bui, 2001 ; Villar-Cocina et al., 2003 ; Zhang et al.,
1996). Les propriétés des mélanges chaux-CBR (ou chaux-CBR
et terre) publiées sont peu nombreuses, partielles et concernent souvent
la résistance en compression. D'une manière
générale, les résistances des mélanges chaux-CBR
augmentent après l'addition de CBR dû à la réaction
pouzzolanique ; bien qu'elles sont encore plus faibles que celles obtenues pour
un ciment de référence (Stroeven et al., 1999 ; Jaturapitakkul et
al., 2003). Les compositions des mélanges chaux CBR trouvées dans
la littérature varient de 20 % à 80 %. Les meilleurs
résultats ont étaient obtenus en utilisant 50% de CBR et 50% de
chaux (Waswa-Sabuni et al., 2003).
| 

