Chapter 2 Literature Survey
2.1 Biology of the malaria parasite
2.1.1 Life cycle of malaria parasite
In humans, malaria is caused by four species of the genus
Plasmodium, namely Plasmonium falciparum, Plasmodium vivax,
Plasmodium ovale and Plasmodium malariae (Wernsdorfer and
McGregor, 1988).
Of these, P. falciparum is the most important as it
causes almost all malaria-associated deaths. There is, however, significant
morbidity associated with P. vivax (Trigg and Kondrachine, 1998). The
biology of P. falciparum is fortunately the best understood of the
four species. The life cycle of P. falciparum is complex and divided
into three overall stages: mosquito, liver and blood stages.
Sexual reproduction of gametocytes occurs in the gut of the
female vector mosquito (Anophele genus) and leads to the formation of zygotes
that bury themselves in the gut lining of the mosquito. These then develop into
oocysts and after some time form sporozoites that migrate to the salivary
glands of the mosquito. When an infected mosquito bites a human host, these
sporozoites enter the blood stream and rapidly make their way to the liver,
invading hepatocytes. During a period of development in the liver of about a
week as tissue schizonts, the parasites multiply asexually, finally
simultaneously rupturing the host cells and entering the blood stream as
merozoites. These merozoites invade red blood cells, entering into the blood
cycle consisting of ring, trophozoite and blood schizont stages as shown in
Figure 1-1.
Asexual reproduction in the blood cell leads to further
merozoites and hence to ever increasing parasitaemia. Some of the merozoites
develop into gametocytes. Upon entering the red cell the gametocytes may be
taken up by mosquitoes to complete the life cycle. Symptoms of the disease
(high fever, headache, malaise, muscle aches...) are entirely associated with
the blood stage and so any curative drug must be specifically active against
this part of the life cycle (Wernsdorfer and McGregor, 1988).
By knowing what is happening in the blood stage of parasite
life cycle, we can understand why, generally speaking, individuals with
abnormal hemoglobin S are resistant to malaria? Normally, about 2% of the
erythrocytes of individuals with sickle -cell anemia are observed to sickle
under low-oxygen concentration conditions found in the capillaries. However,
the lower pH of infected erythrocytes increases the proportion of sickling in
the capillaries up to 40%. Thus during the early stages of malarial infection,
parasite-enhanced sickling probably causes the preferential removal of infected
erythrocytes from the circulation. In the latter stages of infection, when the
parasitized erythrocytes are attached to the capillary walls, the sickling
induced by the low oxygen environment may mechanically and /or metabolically
disrupt the parasite. Consequently, bearers of the sickle cell trait in a
malarial region have an adaptive advantage (Voet and Voet, 1995).
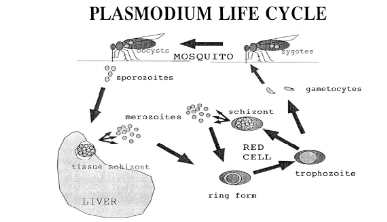
Figure 1-1 A representation of the life cycle
of Plasmodium falciparum. Ring forms, trophozoites and blood schizonts
are collectively referred to as the blood stages of the cycle and are the
specific targets of chloroquine and related antimalarial drugs. After invading
red cells, most merozoites form ring stages and then trophozoites, but a small
fraction instead develop into sexual forms called gametocytes which then
reproduce in the gut of a mosquito when the insect feeds on the infected host.
(Egan et al., 1999).
2.1.2 Hemozoin formation by malaria parasite
During its blood stage, P. falciparum utilises host's
hemoglobin as a food source. This stage occurs in an acidic compartment within
the parasite called a food vacuole that has a pH in the range 5.0-5.6 (Spiller
et al., 2002). Plasmodia degrade hemoglobin and use the amino acids derived
from proteolytic digestion for their biosynthetic requirements. Hemoglobin
degradation is a highly ordered process involving several proteases (Eggleson,
1999; Banerjee, 2002; Rosenthal et al., 2002). Denatured globin formed by the
action of plasmepsins is further degraded into small peptides by other
proteases. A cysteine protease, falcipain, has been characterized from P.
falciparum, which degrades denatured globin (Eggleson, 1999).
Large amounts of free nontoxic heme is released as a product
of hemoglobin degradation (Mavakala and Gushimana, 1991). Released heme from
hemoglobin is autoxidized into ferric form (hematin, hemin or
aquaferriprotoporphyrin IX or H2O-Fe(III)PPIX) that is highly toxic,
inhibiting vacuolar proteases and damaging parasite membranes [Berman and
Adams, 1997]. Detoxification of heme is therefore necessary for the survival
and growth of malaria parasite (Meshnick, 2002).
In the host, detoxification of heme is achieved by an enzyme
called heme oxygenase, which breaks heme to form biliverdin. Another enzyme,
biliverdin reductase, converts biliverdin into bilirubin, which is converted
into a water-soluble conjugate and excreted through urine. Malaria parasite
does not seem to use this pathway for the heme catabolism. Inside the food
vacuole of malaria parasite, heme is converted into hemozoin, popularly known
as malaria pigment. This hemozoin pigment is a dimer of heme units linked
through an iron-carboxylate bond (Pagola et al., 2000).
Pagola et al. have revealed that hemozoin is a hemin
dimer with hydrogen bonding between the dimer units in the crystal as shown in
Figure1-2. In the light of this, the continued use of the word polymer to
describe malaria pigment or -hematin, or the word polymerization to describe
its formation is inappropriate and inaccurate (Pagola et al., 2000; Egan,
2002).
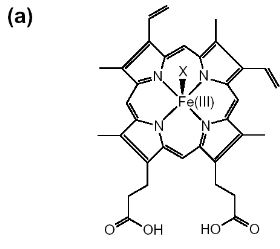
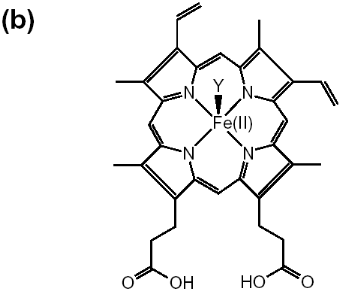
Figure 1-2 Chemical structures are shown for
(a) hematin (aqua or hydroxyferriprotoporphyrin IX),
(b) heme. The dimeric structure for beta-hematin is also
indicated (c). The OH-, H2O group is represented by X in
(a) and the histidine is represented by Y in
(b) (Egan, 2002).
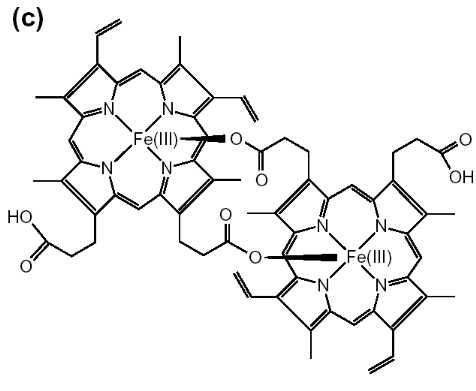
In the dimer, a bond is formed by the linking of central
ferric iron of one heme unit with the propionate side chain of another heme.
This pigment is inert in parasite and released into the host blood supply after
infected erythrocytes burst open at the end of parasite life cycle (Pandey and
Tekwani, 1996). Hemozoin is insoluble in organic solvents (methanol, ethanol,
and acetone) and mildly soluble in alkaline bicarbonate buffer (100 mM, pH
9.0), whereas free heme is soluble in these solvents.
2.2 Some proposed mechanisms of action of antimalarial drugs
Numerous conflicting theories have been put forward over the
past five decades to describe the mechanism of action of antimalarial
drugs. Hypothesis for the mode of action of chloroquine
essentially fall into two broad categories: those in which the drug exerts its
action outside the food vacuole of the parasite and those in which the activity
is located inside the food vacuole.
2.2.1 Mechanism of action of chloroquine and related
antimalarials
2.2.1.1 Extravacuolar mechanisms: DNA
binding
Chloroquine and related drugs exhibit antibacterial activity,
blocking both DNA and RNA synthesis but the required chloroquine concentration
is about one thousand times as much as that needed in curative treatment of
malaria. No binding of mefloquine to DNA has been observed (Slater, 1993; Egan
and Marques, 1999).
2.2.1.2 Intravacuolar mechanisms
Intravacuolar mechanisms seem more plausible because of
substantial accumulation of the drugs in the vacuole. Most
workers in the field currently favour a hypothesis in which quinoline
antimalarial drugs inhibit formation of hemozoin. There is,
however disagreement over how this occurs and there are essentially three
variations of the hypothesis:
1. Slater and Cerami (1992) originally suggested that these
drugs inhibit the putative heme polymerase enzyme.
2. Fitch and Chou (1996) have extended this hypothesis by
suggesting that these drugs are potential regulators of the putative heme
polymerase enzyme.
3. Egan and coworkers (Egan et al., 1994) have shown that
chloroquine, amodiaquine and quinine can directly inhibit formation of
synthetic -hematin and suggested that activity of these drugs in vivo involves
inhibition of hemozoin formation by direct interaction with Fe(III)PPIX. This
hypothesis has also been supported by Dorn and co-workers (Dorn et al., 1995;
Dorn et al., 1998) and further support for this type of mechanism has been
presented by several other laboratories (Sullivan et al., 1996; Basilico et
al., 1997; Hawley et al., 1998) although there are some differences in
detail.
These findings motivated a number of studies on
antimalarial-hematin interactions in both aqueous and non-aqueous solution, as
well as on their interactions with other iron-porphyrins. Many of the earlier
studies concentrated on obtaining visible, Mössbauer and NMR spectroscopic
evidences for hematin-drug interactions and some association constants were
determined. For example, Log K values for the bonding of hemin-drug
are 5.52 (chloroquine), 5.39 (amodiaquine), 4.10 (quinine), 4.04 (9-epiquinine)
and 3.09 (mefloquine), in 40% aqueous DMSO solution, at an apparent pH of 7.5
and 25oC (Egan et al., 1997; Adams et al., 1999). It is clear from
the recent investigation of Egan and coworkers that only 2- and
4-aminoquinolines and their derivatives form strong complexes with Fe(III)PPIX.
Under the conditions of their studies, quinoline, 3-, 5-, 6-, and
8-aminoquinoline, and 4,7-dichloroquinoline exhibited no evidence of
complexation with Fe(III)PPIX. Then, there is a simple correlation between
hemin binding and -hematin inhibitory activity because those of compounds,
which do not form measurable complexes, fail to inhibit -hematin. Surprisingly,
however, not all quinolines, which do form strong complexes with Fe(III)PPIX,
inhibit -hematin formation.(Egan, 2000). Perhaps, they are capable of
inhibiting -hematin formation at high concentration. Egan et al. proposed a
detailed model of the structure-function relationships in chloroquine as
follows:
1. The 4-aminoquinoline nucleus alone provides an hemin
complexing template but is not sufficient for inhibiting the formation of
hemozoin;
2. Introduction of the 7-chloro group is responsible for
inhibition of hemozoin formation but probably has little influence on the
strength of association with hemin;
3. The aminoalkyl side chain is a requirement for strong
antiplasmodial activity. It probably assists in drug accumulation in the food
vacuole. It also appears to enhance the strength of association with hemin in
some cases, but this effect does not appear to be essential for its
activity.
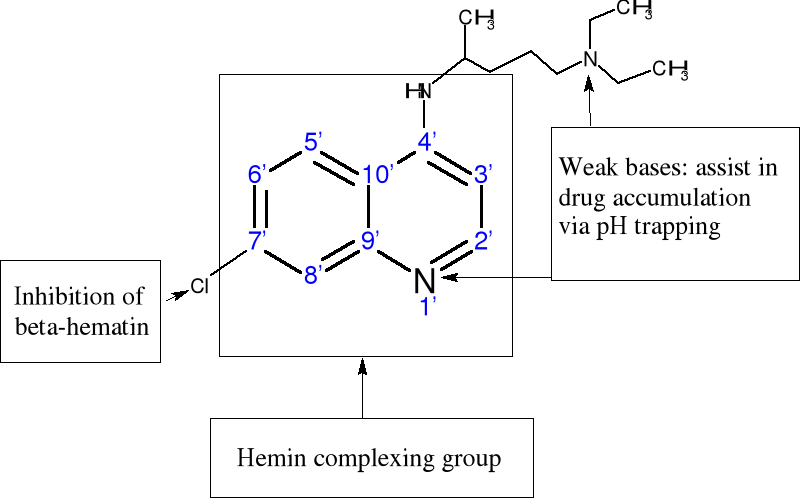
Figure 1-3 Proposed structure-function
relationships in chloroquine based on findings of Egan and coworkers (Egan et
al., 2000).
2.2.1.3 Increased vacuolar pH mechanism
It has been reported that several enzymes like aspartic
proteases, cysteine proteases and metalloproteases (Rosenthal, 1999) are
thought to be involved in the degradation of hemoglobin. Many of these enzymes
are optimally active at pH 4.5-5.0 and it is argued that the food vacuole would
probably need to maintain a similar pH to permit the efficient proteolysis of
hemoglobin (Francis et al., 1997). The work of Homewood and coworkers (Homewood
et al., 1972) in the early 1970s outlined the potential importance of
the pH of the digestive vacuole (pHDV) in the mode of action of
chloroquine (CQ) and similar drugs. CQ is a lipophilic weak base that will pass
through biological membranes in the uncharged form. Once, inside acidic
compartments, CQ is protonated and trapped because the protonated base is
relatively impermeable. If we assume that the digestive vacuole (DV) has a pH
of ~5.0, then this mechanism would permit concentrative uptake of the drug
(Geary et al., 1986). Homewood suggested that CQ might kill parasites by
increasing pHDV, so that the acid proteases of the parasite could no
longer function effectively (Spiller et al., 2002).
2.2.2 Mechanism of action of artemisinin and its
derivatives
2.Artemisinin 3. R= OH Dihydroartemisinin
R= OCH3 Artemether
R= OCH2CH3
Arteether
R=
O2CCH2CH2CO2H Artesunate
1.Deoxyartemisinin
Artemisinin was developed from an ancient Chinese herbal
remedy. Artemisia annua (sweet wormwood or`qinghao') was used by
Chinese herbal medicine practitioners for at least 2000 years. In 1596, Li Shi
zhen, a famous herbalist, recommended it to patients with fever. In 1967,
Chinese scientists screened a series of traditional remedies for drug
activities, and found that extracts of qinghao had potent antimalarial
activity. In 1972, the active ingredient was purified and first named qinghaosu
(essence of qinghao), and then later renamed artemisinin. Western interest in
Artemisinin derivatives (artesunate, artemether, dihydroartemisinin, arteether)
began to grow as multidrug resistant Plasmodium falciparum strains
began to spread. Hundreds of synthetic second generation artemisinin
derivatives and other natural peroxide compounds with good antimalarial
activity have been reported like yingzhaosu, arteflene (Lian et al., 1988;
Hofneiz et al., 1994) as shown in scheme 2-1. Due to their potent antimalarial
activity, fast action, and low toxicity, artemisinin and its derivatives have
distinguished themselves as a new generation of antimalarial drugs. Actually,
it has been established that the dihydroartemisinin combined to the
holotransferrin would be a promising drug against cancer (Singh and Lai,
2001).
4. R=H R1=C6H5
5. .Arteflene
6.Yinzhaosu
R=H R1=CH3
R= OH R1=C6H5
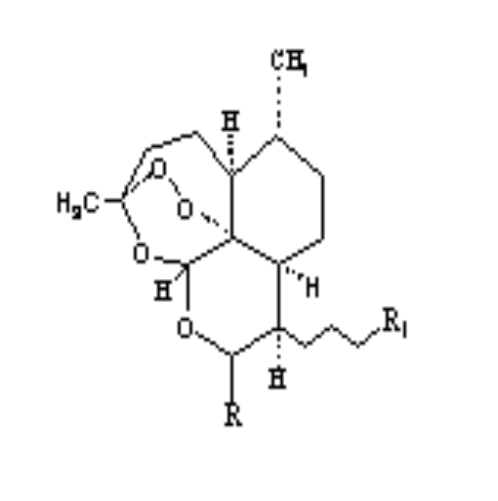
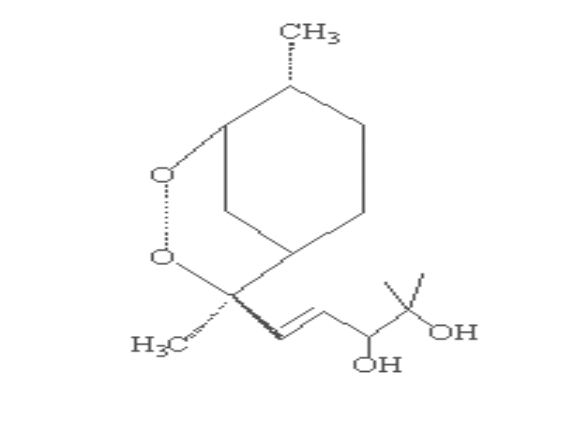
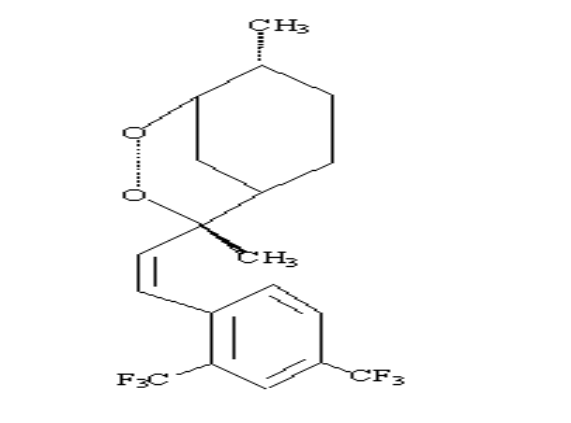
Scheme 2-1 Structures of
artemisinin analogous
The unusual structure of artemisinin molecules might be
indicative of a different mode of action from those of other antimalarial drugs
and hence the high potency against the resistant strains. Although the
mechanism of its antimalarial activity is not clear and still under debate,
there is general agreement that the endoperoxide bridge is essential for the
antimalarial activity of artemisinin since deoxyartemisinin compounds which
lack the endoperoxide moiety are inactive (China cooperative group on
qinghaosu, 1982).
Meshnick et al. proposed a two-step mechanism for
the antimalarial action of endoperoxide:
In the first step, artemisinin is activated by intraparasitic
heme or free Fe (II) ion to produce free toxic carbon-centred radicals,
confirmed by electron paramagnetic resonance (EPR) studies (Meshnick et al.,
1993; Taranto et al., 2002].
In the second step, once formed, the artemisinin-derived free
radicals appear to damage specific intracellular targets, possibly via
alkylation (Berman and Adams, 1997).
But Pandey et al. proposed three possible ways for the effect
of endoperoxide drugs on malaria (Pandey et al., 1999; Kannan et al., 2002):
-Inhibition of hemoglobin degradation
-Inhibition of hemozoin biosynthesis
-Interaction of artemisinin with hemozoin leading to the
breakdown of the hemozoin pigment which could then form a complex with the heme
unity.
These mechanisms are supported by the characterization of a
covalent adduct between artemisinin and heme (Robert and Meunier, 1997) and by
protein alkylation (Meshnick et al., 1991; Yang et al., 1994). Artemisinin also
forms covalent adducts with protein but not with DNA (Yang et al., 1994). Thus,
heme is both an activator and target of the artemisinin derivatives (Posner et
al., 1995).
About the free radicals generated by artemisinin,
there are some controversial discussions on the mechanism of their production.
There is much stronger evidence that carbon-centred free radicals are involved.
In fact, monoelectronic transfer from iron (II) to peroxide resulted in the
cleavage of endoperoxide bond with primary formation of an unstable
oxygen-centred radical, rearrangement and creation of toxic C4-centred free
radicals. It has been proposed that heme attacks the endoperoxide linkage of
artemisinin either at the O1 [Shukla et al., 1995] or O2 position [Tonmumphean,
2001] as shown in scheme 2-2. In pathway A, heme iron attacks the compound at
the O2 position. Later, it rearranges to form C4 free radical. In pathway B,
heme iron attacks the compound at the O1 position after that C3-C4 bond is
cleaved to give carbon radical at C4 as shown in scheme 2.
Thus the presence of heme is necessary for the activation of
artemisinin into an alkylating agent, which preferentially attacks proteins.
The fact that artemisinin becomes cytotoxic in the presence of
ferrous, have triggered some researchers to study its effect on the therapy of
cancer. Since iron influx is high in cancer cells, artemisinin and its
analogous, after incubation with holotransferrin which increase the
concentrations of ferrous iron in cancer cells, selectively kill cancer cells
(Singh and Lai, 2001). In addition to the more largely accepted mechanisms
summarized above, other mechanisms of action have also been proposed. For
example, Jefford proposed that peroxides could interrupt the detoxification
process of heme by transferring an O atom to heme, creating iron-oxene or
oxyheme intermediates, which subsequently disable parasite (Jefford et al.,
1995).
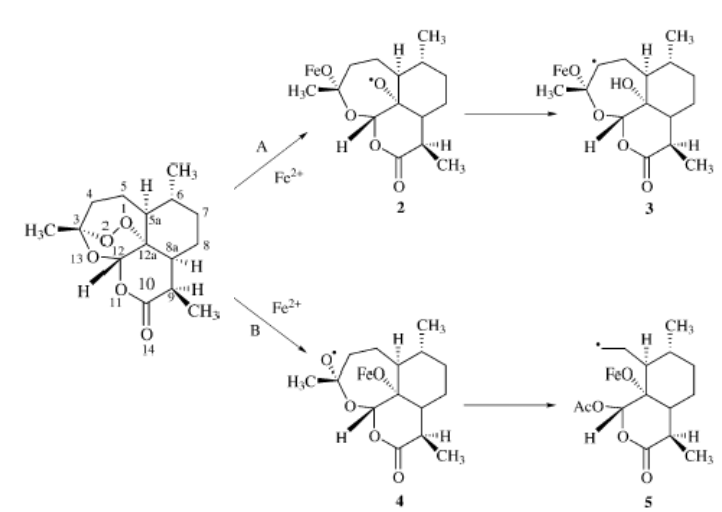
Scheme 2-2
Proposed mechanism of action of artemisinin (Tonmumphean , 200)].
Haynes and co-workers (Haynes et al., 1999) pointed
out that activity is due to the trioxane unity acting as a source of
hydroperoxide, which provides electrophilic oxygenating species, hydroxyl or
alkoxyl radicals via reductive cleavage with Fe (II) or other reducing agents.
These species would be able to hydroxylate biomolecules.
In summary, a schematic diagram of hemoglobin
degradation and related pathways is given in Figure 2-4 (Pandey et al., 1999;
Egan, 2002).
Drug effect 1 :
complex formation with heme
Drug effect 3 :
proteases inhibitors (by endoperoxide only)
Drug effect 2 : inhibition of hemin
dimerization
Drug effect 4 :
Interaction with hemozoin
Toxic effects of heme accumulation :
1. Membrane damage
2. Inhibition of cysteine proteases
| 

