|
RÉPUBLIQUE ALGÉRIENNE DÉMOCRATIQUE ET
POPULAIRE
MINISTÈRE DE L'ENSEIGNEMENT SUPÉRIEUR ET DE LA
RECHERCHE
SCIENTIFIQUE

Université Abderrahmane
MIRA-Bejaïa
Faculté des Sciences Exactes
Département
de Chimie
MÉMOIRE
Présenté
par
M. AZOUZ L'hachemi
En vue de l'obtention du
diplôme de
MAGISTER
En Chimie
Option : Chimie
de l'environnement
Étude des interactions de mélanges
(polymères
biodégradables/principe actif) obtenus
par différentes méthodes de
préparations
Soutenu le 14/04/2011 à 9H00
Devant le jury composé de :
|
M. M. BERKANI
|
Maître de conférences A
|
U. A. M.
|
Bejaïa
|
Président
|
|
M. H. DJIDJELLI
|
Professeur
|
U. A. M.
|
Bejaïa
|
Examinateur
|
|
M. N. BEZZI
|
Maître de conférences A
|
U. A. M.
|
Bejaïa
|
Examinateur
|
|
M. F. REZGUI
|
Maître de conférences A
|
U. A. M.
|
Bejaïa
|
Rapporteur
|
A mon dieu le miséricordieux
A tous ceux qui travaillent pour guider l'humanité
vers le chemin de la Vérité A la mémoire de mon
père
A ma mère
A mes soeurs et mes frères
Remerciements
Remerciements
Ce travail a été réalisé au
laboratoire des Matériaux Organiques et au laboratoire de Génie
des Procédés et Pharmaceutique à l'université
A/Mira - BEJAIA.
Je tiens tout d'abord à remercier le directeur de ce
mémoire, M. REZGUI Farouk (maître de conférences à
l'université A/Mira - Bejaïa), pour m'avoir fait confiance et
engagé afin de travailler sur ce sujet, puis pour m'avoir guidé,
conseillé, tout en me laissant une grande liberté. Je le remercie
aussi pour sa gentillesse, sa disponibilité et ses qualités
humaines. Je
suis très heureux de l'avoir eu comme directeur de
mémoire.
J'exprime toute ma reconnaissance à M. BERKANI Madjid,
pour avoir présidé admirablement le jury de ce mémoire
ainsi que pour sa bonne humeur de tous les jours.
J'adresse mes remerciements aux messieurs M. BEZZI Nacer et M.
DJIDJELLI Hocine pour avoir acceptés d'être examinateurs et pour
l'honneur qu'ils me font de participer au jury.
Je tiens à remercier mes collègues de la
promotion avec qui j'ai passé de très bons moments. Sans oublier
également tous les techniciens des laboratoires qui ont contribué
à l'accomplissement de ces travaux.
Enfin, je terminerai en remerciant mes proches : ma mère,
ma grande mère, mes soeurs, mes frères et mes amis qui m'ont
toujours aidé, soutenu et supporté durant ces années.
A vous tous, merci.
SOMMAIRE
Introduction générale 1
SECTION BIBLIOGRAPHIQUE
Chapitre
I. Rappel bibliographique sur le poly(acide lactique)
et
l'ibuprofène
Partie A
5
5
7
7
8
9
9
10
11
11
12
13
15
15
16
17
17
18
20
21
21
22
23
24
28
28
29
A-I. Poly(acide lactique) (PLA) .
A-I. 1. Le monomère de base de poly(acide lactique)
A-I.2. Méthodes de synthèse de poly(acide lactique)
.
A-I.2. 1. Polymérisation par condensation directe en
absence de catalyseur ..
A-I.2.2. Polymérisation par condensation directe en
présence de catalyseur ...
A-I.2.2. 1. Polymérisation par condensation
azéotropique .
A-I.2.2.2. Polymérisation à l'état solide
.
A-I.2.3. Polymérisation par ouverture de cycle ..
A-I.2.3. 1. Polymérisation anionique ...
A-I.2.3 .2. Polymérisation cationique ..
A-I.2. 3.3. Mécanisme de coordination-insertion ...
A-I.3. Catalyseurs utilisés dans la synthèse de PLA
..
A-I.4. Propriétés de poly(acide lactique) ..
A-I.4. 1. Propriétés thermiques ..
A-I.4.2. Propriétés mécaniques ..
A-I. 5. Vieillissement et biodégradation de poly(acide
lactique) .
A-I. 5.1. Vieillissement physique de poly(acide lactique) ...
A-I.5.2. Dégradation non-biologique (ou abiotique) de
poly(acide lactique) ..
A-I.5.3. Biodégradation de poly(acide lactique)
A-I.5.4. Facteurs influençant la biodégradation de
poly(acide lactique)
A-I.5.5. Dégradation biologique (ou biodégradation)
de poly(acide lactique) .
A-I.5.5.1. Dégradation microbienne de poly(acide lactique)
.
A-I.5.5.2. Dégradation enzymatique de poly(acide lactique)
.
A-I.6. Production industrielle durable de poly(acide lactique)
...
Partie B
B-I. Ibuprofène
B-I. 1. Identification de l'ibuprofène
B-I.2. Chimie médicinale de l'ibuprofène
B-I.2. 1. Métabolites de l'ibuprofène 29
B-I.2.2. Enantiomères de l'ibuprofène 30
B-I.3. Pharmacie de l'ibuprofène 30
B-I. 3.1. Caractéristiques de l'ibuprofène
30
B-I. 3.1.1. Caractéristiques physiques et chimiques de
l'ibuprofène 30
B-I. 3.1.2. Caractéristiques physicochimiques de
l'ibuprofène 32
B-I. 3.1.3. Caractéristiques cristallographiques de
l'ibuprofène .. 32
B-I.3.2. Disponibilité de l'ibuprofène dans le
monde 33
B-I.4. Pharmacocinétique de l'ibuprofène 34
B-I.4. 1. Mécanisme d'action 34
B-I.4.1. Absorption de la molécule d'ibuprofène
dans l'organisme 35
B-I.4.2. Distribution de la molécule d'ibuprofène
dans l'organisme 35
B-I.4.3. Demi-vie et élimination de l'ibuprofène
dans l'organisme 36
B-I.5. Utilisation thérapeutique de l'ibuprofène ..
36
B-I.6. Effets indésirables et intoxication de
l'ibuprofène 36
B-I.7. Utilisation de l'ibuprofène dans les
systèmes à libération prolongée 37
Chapitre II. Vectorisation de principes actifs
II.1. Historique...... ... ... ... ... ... ... ...40
II.2. Concept général de la vectorisation de
principes actifs 41
II.3. Différentes voies d'administration des
médicaments 43
II.3.1. Voie transdermale 43
II.3.2. Voie oral 44
II.3.3. Voie nasale 46
II.3.4. Voie pulmonaire 46
II.4. Utilisation des polymères biodégradables
pour la libération contrôlée de principes
actif 48
II.4.1. Différents types de profiles de la vectorisation
de principes actifs 48
II.4.2. Différents systèmes utilisés pour la
vectorisation de principes actifs 49
II.4.2.1. Systèmes à diffusion
contrôlée 49
a/ Système réservoir (membrane) 49
b/ Système matriciel (monolithique) . 50
II.4.2.2. Systèmes chimiquement contrôlés
50
a/ Systèmes bioérodibles et biodégradables
.. 50
b/ Systèmes à chaînes pendants 51
II.4.2.3. Systèmes à solvants activés .
52
a/ Système à gonflement contrôlé .
52
b/ Systèmes osmotiquement contrôlés
......53
II.4.3. Différents types de vecteurs de principes actifs
et leurs architectures ..54
II.4.3.1. Architecture des polymères . 54
a/ Polymère linéaire 54
b/ Polymères branchés 55
c/ Polymères réticulés 56
II.4.3.2. Différents types de vecteurs de principes
actifs 57
II.4.3.2.1. Micelles polymériques 58
II.4.3.2.2. Vésicules polymériques (ou
polymèrsomes) 60
61
63
64
65
66
66
68
70
71
73
II.4.3.2.3. Micro et nanoparticules ...
II.4.3.2.4. Hydrogels .
II.5. Application de poly(acide lactique) dans le domaine
pharmaceutique
II.5.1. Rôle de l'interaction polymère/principe
actif dans la libération prolongée de principes actifs
..
II.5.2. Quelques exemples d'application de poly(D,L-acide lactique) dans
le domaine
de la vectorisation de principes actifs
II.5.2.1. Chimiothérapie du cancer
II.5.2.1.1. Utilisation du PDLLA pour améliorer
l'efficacité thérapeutique de l'agent anticancéreux
bléomycine (BLM)
II.5.2.1.2. Ciblage d'un agent anticancéreux
hydrophobe (Paclitaxel) par l'utilisation
d'un copolymère amphiphile (PVP-b-PDLLA) ..
II.5.2.2.
Application de poly(D,L-acide lactique) dans le domaine de
l'antibiothérapie .
II.5.2.3. Application dermato-thérapeutique de
poly(D,L-acide lactique) ..
SECTION EXPÉRIMENTALE
Chapitre III. Matériaux et méthodes
utilisées
Partie I. Synthèse de poly(D,L-acide
lactique) par la méthode de
polycondensation
azéotropique
I.1.
76
76
76
77
Introduction
I.2. Matériaux utilisés
I.3. Protocole expérimental
I.4. Mesure de la masse viscosimétrique des
polymères obtenus Partie II. Préparation des
formulations polymère/principe actif
I.1. Introduction ... ... ... ... ...... ... 78
I.2. Matériaux utilisés ... ... ... ... ......
... 78
I.3. Protocoles expérimentaux 78
II.3.1. Préparation des mélanges physiques PDLLA/IB
... 79
II.3.2. Préparation des mélanges IB/PDLLA par
évaporation de solvant
II.3.3. Préparation des mélanges IB/PDLLA par
fusion à chaud 79
79
II.4. Techniques de caractérisation des formulations
80
II.4.1. Analyse thermique 80
II.4.1.1. Analyse thermique ATG - ATD . 80
II.4.1.2. Analyse thermique différentielle 80
II.4.2. Analyse spectrale 80
II.4.1.1. Spectrométrie infrarouge à
transformé de Fourier (IRTF) 80
II.4.2.2. Diffraction des rayons X
II.4.2.3. Microscopie électronique à balayage
(MEB) .. 81
81
II.4.2.4. Spectrophotométrie UV-Visible . 81
II.5. Etude de dissolution in vitro des échantillons
81
II.5.1. Préparation du milieu tampon
II.5.2. Courbes d'étalonnage de l'ibuprofène
à chaque valeur de pH 81
83
83
II.5.3. Préparation des comprimés
II.4.4.4. Test de dissolution
Chapitre IV. Résultats et discussions
Partie I. Synthèse et caractérisation de
poly(D,L-acide lactique)
I.1.
84
86
86
86
87
89
89
89
Synthèse de PDLLA
I.2. Caractérisation des polymères obtenus
I.2.1. Analyse thermique
I.2.1.1. Etude TG/DTG
I.2.1.2. Etude DSC
I.2.2. Analyse structurale
I.2.2.1. Spectres IRTF des polymères obtenus
I.2.2.2.
Spectres DRX des PDLLA obtenus
Partie II. Caractérisation du principe actif
(Ibuprofène pur)
II.1. 91
91
91
92
92
93
Analyse thermique ... ... ... ... ......
II.1.1. Etude TG/DTG
II.1.2. Analyse DSC
II.2. Analyse structurale ...
II.2.1. Spectre IRTF de l'ibuprofène
II.2.2. Spectre DRX de l'ibuprofène
.
Partie III. Etude et caractérisation des
différents mélanges PDLLA/IB
III.1. Microscopie électronique à balayage (MEB)
94
95
98 104 104 107 109
112
113
117
120
III.2. Spectroscopie infrarouge à transformé de
fourrier (IRTF)
III.3. Analyse cristallographique des différents
échantillions
III.4. Tests de dissolution in vitro de l'ibuprofène
III.4.1. Effet du taux d'enrobage
III.4.2. Effet de la masse moléculaire de la matrice PDLLA
III.4.3. Effet du pH du milieu du dissolution
III.4.4. Effet de la méthode de préparation des
mélanges
III.5. Interactions ibuprofène/poly(D,L-acide lactique)
Conclusion générale ..
Références bibliographiques .
LISTE DES FIGURES
6
6
7
8
9
10
10
11
12
13
14
15
19
20
22
26
28
29
29
30
31
33
35
36
42
43
43
44
47
48
49
50
51
52
53
54
55
Figure 1. Production de l'acide lactique à partir des
ressources renouvelables
Figure 2. Différents isomères optiques de l'acide
lactique .
Figure 3. méthodes de synthèse du PLA ..
Figure 4. Synthèse de PLA par polycondensation direct .
Figure 5. Polymérisation à l'état solide
Figure 6. Synthèse de PLA par polymérisation par
ouverture de cycle
Figure 7. Les différents isomères du lactide
Figure 8. Polymérisation par ouverture de cycle anionique
Figure 9. Polymérisation par ouverture de cycle cationique
Figure 10. Mécanisme de coordination-insertion
Figure 11. Octoate d'étain
Figure 12. Polymérisation de lactide par le
mécanisme coordination-insertion catalysé
par l'octoate d'étain .
Figure 13. Hydrolyse de poly(acide lactique) dans un milieu
alcalin ..
Figure 14. Hydrolyse de poly(acide lactique) dans un milieu acide
Figure 15. Mécanisme de dégradation de poly(acide
lactique)
Figure 16. Processus de préparation sans solvant de
poly(acide lactique) ..
Figure 17. Structure de l'ibuprofène
Figure 18. La structure des deux principaux métabolites de
l'ibuprofène
Figure 19. la structure des deux autres métabolites de
l'IB .
Figure 20. La structure des deux formes
énantiomèriques de l'IB ..
Figure 21. Solubilité de l'ibuprofène en fonction
du pH .
Figure 22. Formation d'un dimère cyclique entre deux
molécules d'IB .
Figure 23. Conversion de R(?)-ibuprofène en
S(+)-ibuprofène au moyen d'une activité catalytique de l'acyl
coenzyme thioestérase
Figure 24. Profiles d'absorptions cumulatifs
moyens des énantiomères de l'IB après
une administration séparée de l'IB racémique
et de chaque énantiomère
Figure 25. Plage
thérapeutique d'un principe actif : Application par un système
à libération contrôlée de principes actifs
comparé à un système d'administration par
injection .
Figure 26. Différentes voies d'administration
Figure 27. Processus de perméation transdermale
Figure 28. Anatomie de la partie gastro-intestinal humaine
Figure 29. Différentes régions de la partie
respiratoire humaine .
Figure 30. Différents types de profiles de
libération de principes actifs .
Figure 31. Libération d'un principe actif à partir
d'un système réservoir : (a) système implantable ou oral,
(b) système transdermale .
Figure 32. Libération d'un principe
actif à partir d'un système matriciel à
libération
contrôlée
Figure 33. Libération du
principe actif à partir des systèmes biodégradables :
(a)
érosion volumique, (b) érosion superficielle
Figure 34. Clivage de squelette polymère .
Figure 35. Libération du principe actif à partir
des systèmes à gonflement contrôlé : (a)
système réservoir, (b) système matriciel
Figure 36. Pompe osmotique
Figure 37. Modèle de Ringsdorf pour le conjugué
polymère-principe actif .
55
56
56
58
59
60
61
62
63
64
67 69 69 71 73 75
77
79
80
82
82
83
87
88
89
90
91
Figure 38. Différentes formes architecturales des
polymères linéaires
Figure 39. Différentes formes architecturales des
polymères branchés
Figure 40. Différentes formes architecturales des
polymères réticulés
Figure 41. Vecteurs pharmaceutiques
Figure 42. Trois principaux types de micelles a base de block
copolymère linéaire (a) micelle de block copolymère
commun, (b) micelle de conjugué block copolymèreprincipe actif,
(c) micelle de complexe block ionomère
Figure 43. Procédures
de chargement de principes actifs : (A) équilibre simple, (B)
dialyse, (C) émulsion o/w, (D) solution casting, et (E)
lyophilisation ..
Figure 44. Formation des vésicules a base des
copolymères di- et triblock ..
Figure 45. Les deux modèles de particules utilisées
en libération contrôlée .
Figure 46. Libération de principes actifs a partir
d'hydrogels sensibles aux stimuli et
intelligents
Figure 47. Application de poly(acide lactique) .
Figure 48.
Les stratégies de ciblage d'un principe actif (PA) anticancéreux.
(A) libération du PA tant au niveau des tissus tumoraux que les tissus
sains, (B) libération du PA dans la tumeur et internalisation du PA par
les cellules tumorales, (C) internalisation du vecteur suite a l'interaction
spécifique entre la molécule de reconnaissance attachée a
la surface du vecteur et son récepteur sur la membrane des cellules
cibles, (D) libération du PA en réponse a l'environnement ou a un
stimulus
externe
Figure 49. Libération de la bléomycine
a partir de BLM-PDLLA dans le milieu
physiologique (pH 7,4 a 37°C) .
Figure 50. Variation de
la concentration de BLM dans les tissus sous-cutanés autour de
site d'implantation
Figure 51. Cinétique de
libération de paclitaxel a partir des nanoparticules de PDLLA
préparées dans le dichlorométhane comme
solvant organique .
Figure 52. Profils de libération de gentamycine a
partir de l'acier inoxydable recouvert
de PDLLA ..
Figure 53. Libération in vitro de nile
rouge (NR) et coumarine-6 (Coum-6) a partir des
particules de PDLLA dans un système biphasique
(hexane/solution tampon) .
Figure 54. Montage de polymérisation
azéotropique de D,L-acide lactique dans une
atmosphère inerte ..
Figure 55. Montage de la méthode d'évaporation de
solvant sous vide
Figure 56. Montage de la méthode de fusion a chaud
Figure 57. Courbe d'étalonnage de l'ibuprofène a pH
7,4
Figure 58. Courbe d'étalonnage de l'ibuprofène a pH
5,8
Figure 59. Représentation schématique d'une cellule
de dissolutest
Figure 60. Courbes de TG/DTG obtenus a 10 °C/min sous
atmosphère d'azote pour le
poly(D,L - acide lactique) de différentes masses
moléculaires
Figure 61. Les thermogrammes DSC des trois
polymères PDLLA obtenus a une vitesse
de chauffage de 10 °C/min
Figure 62. Les spectres IRTF
des trois polymères PDLLA : (a) PDLLA 1000, (b)
PDLLA 3000 et (c) PDLLA 9000 Da) .
Figure 63. Les spectres DRX
de poly(D,L-acide lactique) : (a) PDLLA 1000, (b)
PDLLA 3000 et (c) PDLLA 9000 Da
Figure 64. Courbe de TG/DTG
obtenu a 10 °C/min sous atmosphère d'azote pour
l'ibuprofène pur
Figure 65. Courbe DSC de l'ibuprofène pur .
92
93
94
95
96
96
97
98
99
100
101
101
102
102
105
106
107
108
109
Figure 66. Spectre IRTF de l'ibuprofène pur
Figure 67. Spectre DRX de l'ibuproféne pur
Figure 68. Micrographies électronique à balayage :
(a) Ibuprofène, (b) Poly(D,L-acide lactique), (c) Mélanges
physiques, (d) Mélanges en fusion et (e) Mélanges par
évaporation de solvant ...
Figure 69. Spectre IRTF des produits purs
(a) ibuprofène pur, (b) PDLLA 3000 pur, et des mélanges physiques
(c) IB+5%PDLLA, (d) IB+15%PDLLA et (e)
IB+25%PDLLA .
Figure 70. Spectre IRTF des produits purs (a)
ibuprofène pur, (b) PDLLA 3000 pur, et
des mélanges à fusion (c) IB+5%PDLLA, (d)
IB+15%PDLLA et (e) IB+25%PDLLA
Figure 71. Spectre IRTF des produits purs
(a) ibuprofène pur, (b) PDLLA 3000 pur, et des mélanges par
évaporation de solvant (c) IB+5%PDLLA, (d) IB+15%PDLLA et (e)
IB+25%PDLLA
Figure 72. Spectre IR-TF dans la région
spectrale du carbonyle. (A) Mélanges physiques, (B) Mélanges
à fusion et (C) Mélanges par évaporation de solvant de IB
et PDLLA3000. (a) IB, (b) PDLLA, (c) IB+5%PDLLA, (d) IB+15%PDLLA et (e)
IB+25%PDLLA .
Figure 73. Diffraction des rayons X sur poudre
de (a) PDLLA3000, (b) IB pur, (c) IB+5%PDLLA3000 (F5), (d) IB+15%PDLLA3000
(F15) et (e) IB+25%PDLLA3000
(F25) ..
Figure 74. Diffraction des rayons X sur poudre des
produits purs (IB et PDLLA3000) et de la formulation F25 (mélange
physique) dans les intervalles des angles 2è de (1) 5,0°-
7,0°, (2) 11,5°-13,0°, (3) 16,0°-17,2°
et (4) 19,5°-23,0°
Figure 75. Diffraction des rayons X sur poudre
de (a) PDLLA3000, (b) IB pur, (c) IB+5%PDLLA3000 (F5), (d) IB+15%PDLLA3000
(F15) et (e) IB+25%PDLLA3000
(F25)
Figure 76. Diffraction des rayons X sur poudre des
produits purs (IB et PDLLA3000) et de la formulation F25 (mélange de
fusion à chaud) dans les intervalles des angles 2è de
(1) 5,0°-7,0°, (2) 11,5°-13,0°, (3)
16,0°-17,2° et (4) 19,5°-23,0° ..
Figure 77. Diffraction
des rayons X sur poudre de (a) PDLLA3000, (b) IB pur, (c) IB+5%PDLLA3000 (F5),
(d) IB+15%PDLLA3000 (F15) et (e) IB+25%PDLLA3000
(F25)
Figure 78. Diffraction des rayons X sur poudre des
produits purs (IB et PDLLA3000) et de la formulation F25 (mélange par
évaporation de solvant) dans les intervalles des
angles 2è de (1) 5,0°-7,0°, (2)
11,5°-13,0°, (3) 16,0°-17,2° et (4) 19,5°-23,0°
.
Figure 79. Effet du taux d'enrobage sur les cinétiques de
libération de l'IB à partir des microgranules de PDLLA à
pH 7,4. (a) mélanges physiques, (b) mélanges en fusion et
(c) mélanges par évaporation de solvant
Figure
80. Effet du taux d'enrobage sur les cinétiques de libération de
l'IB à partir des microgranules de PDLLA à pH 5,8. (a)
mélanges physiques, (b) mélanges en fusion et
(c) mélanges par évaporation de solvant
Figure
81. Effet de la masse moléculaire de PDLLA sur les cinétiques de
libération de l'IB à pH 7,4. (a) mélanges physiques, (b)
mélanges en fusion et (c) mélanges par
évaporation de solvant
Figure 82. Effet de la masse
moléculaire de PDLLA sur les cinétiques de libération de
l'IB à pH 5,8. (a) mélanges physiques, (b) mélanges en
fusion et (c) mélanges par
évaporation de solvant
Figure 83. Effet du pH sur la cinétique de
libération de l'ibuprofène pur
Figure 84. Effet du pH sur la cinétique de
libération de l'IB à partir de la formulation
110
110
111
112
115
F5 : (a) mélange physique, (b) mélange en fusion et
(c) mélange par évaporation de solvant
Figure 85. Effet du pH
sur la cinétique de libération de l'IB à partir de la
formulation F15 : (a) mélange physique, (b) mélange en fusion et
(c) mélange par évaporation de
solvant
Figure 86. Effet du pH sur la cinétique de
libération de l'IB à partir de la formulation F25 : (a)
mélange physique, (b) mélange en fusion et (c) mélange par
évaporation de
solvant
Figure 87. Effet de la méthode
préparation des mélanges PDLLA/IB sur les profils de
libération de l'IB à partir de microgranules enrobés
à différents taux d'enrobage (5, 15
et 25%) en tampon phosphate de pH 7,4 à 37°C
Figure 88. Schéma représentatif d'éventuelles
interactions dans les dispersions solides IB/PDLLA. (a) dimère
symétrique d'IB, (b) PDLLA seul et (c) IB liée à la
chaîne de
PDLLA par une liaison hydrogène
LISTE DES ABRÉVIATIONS ET SYMBOLES
Les
abréviations
Polymères
PLA : Poly(acide lactique)
L-PLA (ou PLLA) : Poly(L-acide lactique) D-PLA (ou PDLA) :
Poly(D-acide lactique)
DL-PLA (ou PDLLA) : Poly(D,L-acide lactique)
PCL : PolyCaproLactone
PPC : PolyPropylène Carbonate
PE : PolyEthylène
PS : PolyStyrène
PP : PolyPropylène
PVP : Polyvinylpyrrolidone
PET : Polyéthylènetériphtalate
PLGA : Poly(acide lactide-co-glycolide) MPEG : Méthoxy
polyéthylène glycol HPMC : Hydroxypropylméthylcellulose
Ddex : DEAE dextran
Principes actifs
AINS : Anti-Inflammatoire Non Stéroïdiens
IB : Ibuprofène PTX : Paclitaxel BLM :
Bléomycine
BLM-SOL : Solution de la bléomycine
Autres :
CoA : Coenzyme A
pH : Potentiel Hydrogène Sn(Oct)2 : Octoate
d'étain
USA : Etats Unis d'Amérique
UK : Royaume-Uni
Drug ou PA : Principe actif
DSC : Analyse enthalpique différentielle COX :
cyclo-oxygénase
PEGylation : conjugué polyéthylène
glycol-principe actif
ADN : Acide désoxyribonucléique
UV : Ultraviolet
ATR : Cristal à réflexion totale
atténuée IR-TF : Infrarouge à transformée de
fourrier
CrEL : Huile de recin polyoxyélhylée DRX :
Diffraction des rayons X
MEB : Microscopie électronique à balayage
ATD : Analyse différentielle thermique ATG : Analyse
thermogravimétrique LMO : Laboratoire des matériaux organiques
DMF : N,N-diméthylformamide
Les symboles
Caractères grecques :
ëmax : Longueur d'onde maximale ?r :
Viscosité relative
?sp : Viscosité spécifique
[?] : Viscosité intrinsèque
í : Fréquence
Caractères latins :
Mw : Masse Moléculaire Moyenne en poids Tf :
Température de fusion
Tg : Température de transition vitreuse Td :
Température de décomposition
Mv : Masse moléculaire moyenne
viscosimétrique
K : Kelvin
MPa : Millipascale mg : milligramme ug/g : Microgramme par
gramme
um : Micromètre h : Heure
cfu : colony-forming unit
m : mètre
cm : Centimètre
nm : Nanomètre
um : Micromètre
mmHg : Millimètre mercure
KV : Kilovolt
mA : Milliampère min : Minute
l : Litre
ml : Millilitre
mW : Milliwatt
cps : Cycle par seconde
Å : Angström
mol : Mole
R : Rectus (ou droit)
S : Sinister (ou gauche)
pKa : Constante d'acidité
ÄHfus : Enthalpie de fusion
ÄHsub : Enthalpie de sublimation ÄHvap : Enthalpie
de vaporisation Z : Nombre de motifs par maille
tmax : Temps de la concentration maximale Cmax : Concentration
maximale de plasma t1/2 : Période de demi-vie
t : Temps
C : Concentration
Tc : Température de cristallisation
Xc : Taux de cristallinité
dm/dt : Différentielle de la masse M : Masse
ÄHf : Enthalpie de fusion
Cp : Capacité calorifique
Unités de mesure :
KJmol-1 : Kilojoule par mole
JK-1g-1 : Joule par Kelvin par gramme
Liste des tableaux
LISTE DES TABLEAUX
Tableau 1. Propriétés thermiques typiques de
poly(acide lactique) . 16
Tableau 2. Propriétés mécaniques typiques de
poly(acide lactique) 17
Tableau 3. Les microorganismes responsables de la
dégradation de PLA .. 24
Tableau 4. Les principaux acteurs de la production de PLA
à travers le monde . 25
Tableau 5. Les différents impuretés
détectées dans l'ibuprofène 31
Tableau 6. Solubilité de l'ibuprofène dans des
solvants organiques 31
Tableau 7. Caractéristiques physiques de
l'ibuprofène 32
Tableau 8. Caractéristiques chimiques de
l'ibuprofène 32
Tableau 9. Quelques propriétés physicochimiques de
l'IB .. 32
Tableau 10. Donnée cristallographiques sur la maille
d'ibuprofène 33
Tableau 11. Disponibilité des formes galéniques
dans l'Europe et les Etats-Unis (%) 34
Tableau 12. Physiologie de la partie gastro-intestinale humaine .
45
Tableau 13. Méthodes de synthèse des
différents types de nano et microparticules 62
Tableau 14. Constitution des formulations préparées
.. 79
Tableau 15. Constitution des comprimés . 83
Tableau 16. Les masses viscosimétriques de PLA pour chaque
temps de
polymérisation .. 84
Tableau 17. Les différents informations sur le PDLLA
données par DSC .. 88
Tableau 18. Les bandes d'absorption infrarouge
caractéristiques de l'ibuprofène pur... 92 Tableau 19.
Diffraction des rayons X des différentes formulations de
l'ibuprofène
avec le PDLLA 3000 103
Liste des schémas
LISTE DES SCHÉMAS
Schéma 1. Processus de biodégradation des
polymères biodégradables . 21
Schéma 2. Représentation de la liaison ester entre
la chaîne polymère et le principe
actif (Acrylique/IB) .. 65
Schéma 3. Possibilité
d'une liaison électrostatique entre le polymère et le principe
actif (DEAE/IB) 66
Schéma 4. Possibilité d'une hydrogène entre
le polymère et le principe actif (PVP/IB) 66
Schéma 5. Réaction de polymérisation par
polycondensation azéotropique 98
Schéma 6. Mécanisme réactionnel de la
synthèse de PLA en présence d'un catalyseur (SnCl2) . 100
INTRODUCTION GÉNÉRALE
Introduction générale
Pendant les soixante dernières années, les
matériaux polymères synthétiques issus essentiellement de
la pétrochimie ont été développés
progressivement, avec une production mondiale d'environ 140 millions de tonnes
par an. Durant cette période, les matériaux polymères ont
envahi notre univers quotidien, on les retrouvent dans de divers domaines
à savoir, l'emballage, le bâtiment, le transport, les
équipements électriques et électroniques, l'ameublement et
la décoration, le loisir...etc. Ce succès est dû
principalement à leur faible coût, reproductibilité
à grande vitesse, excellentes propriétés mécaniques
et leur durabilité (grande résistance au vieillissement et aux
attaques biologiques). Cependant, le développement et l'exploitation
intense de la matière plastique pour des usages courants, se sont
traduits par l'accumulation de déchets non biodégradables,
à durée de vie très longue, dans l'environnement. Ceci a
provoqué une véritable source de nuisance visuelle,
d'encombrement des décharges et de pollution des sols et des eaux
(M. Vert, 2002 ; E. Rudnik, 2008 ; A. A. Shah, 2008).
Suite à cette situation et face à la hausse du
prix du pétrole et à la diminution progressive des stocks,
l'industrie plastique s'est orientée vers une alternative aux
matières premières conventionnelles. Plusieurs solutions ont
été envisagées pour réduire l'impact de ces
matériaux sur l'environnement. La première est le recyclage
chimique ou physique pour donner une nouvelle vie à ces
polymères. La deuxième est l'incinération en les utilisant
comme combustibles afin de produire de l'énergie (i.e. valorisation de
la matière plastique). Cependant, le recyclage et la
réutilisation des matières plastiques sont limitées par la
complexité des résidus rejetés et les coûts
élevés. La valorisation quant à elle se confronte à
la nécessité du retraitement des fumés, en particulier
pour éliminer les gaz à effet de serre (NOx,
SOy ...) (M. Vert, 2002 ; E. Chiellini,
2001).
Le respect de l'environnement est un point capital dans le
contexte du développement durable. L'homme doit agir de cette
façon pour préserver les ressources fossiles et réduire la
pollution de la terre. La fabrication des produits industriels doit consommer
moins d'énergie et la matière première doit être en
priorité issue de ressources renouvelables, en particulier du monde
végétal (N. Lucas).
Par leur abondance et leur diversité, les
polymères issus du monde végétal offrent une nouvelle
source de matières premières renouvelable pour l'industrie
plastique. Grâce à leur biodégradabilité, ces
polymères pourraient constituer une solution aux problèmes
environnementaux engendrés par les importants tonnages de déchets
plastiques (H. N. Rabetafika,
2006). En plus, ces polymères issus de ressources renouvelables
peuvent être éliminés simplement par biodégradation
(ex. compostage) après utilisation (C. Bastioli,
2005).
Généralement, les polymères issus de
ressources renouvelables peuvent être classifiés en trois groupes
:
- polymères naturels comme amidon, protéine, et
cellulose;
- polymères issus de la fermentation microbienne comme
polyhydroxybutyrate (PHB);
- polymères synthétiques à partir de
monomères naturels comme poly(acide lactique (PLA). Suite à leurs
propriétés comparables aux polymères conventionnels, leur
biocompatibilité et
leur biorésorbabilité, les polymères
synthétiques à partir de monomères issus de ressources
renouvelables ont attirés l'attention des scientifiques durant les deux
dernières décennies. Durant les dix dernières
années, ces polymères ont été
considérés comme les matériaux les plus utilisés
tant d'un point de vue académique (i.e. recherches aux laboratoires)
qu'industriel
(E. Rudnik, 2008 ; L. Yu, 2009).
Parmi les polymères synthétiques
biodégradables, le poly(acide lactique) (PLA) est apparu comme un
candidat très prometteur utilisé dans divers domaines
d'application à savoir la médecine, l'agriculture et
l'emballage.... Le poly(acide lactique) est un polyester aliphatique
thermoplastique dérivé 100 % de ressources renouvelables tel le
maïs. Grâce à sa durabilité, sa
biodégradabilité, sa transparence et ses propriétés
mécaniques, la production du PLA n'a cessé de croître
(L. Yu, 2009 ; R. M. Rasal, 2010).
Comme son monomère de base est issu d'un
métabolite naturel du corps vivant, le poly(acide lactique) est un
polymère bien toléré par l'organisme et ne présente
aucune toxicité, ce qui a permis une large utilisation dans le domaine
médicale et en particulier, pharmaceutique en tant que vecteurs de
principes actifs de part la capacité d'encapsuler, de transporter et de
libérer certains molécules insolubles dans les milieux aqueux
(H. Rabetafika, 2006).
Actuellement, l'industrie pharmaceutique recherche de
nouvelles formes médicamenteuses, libérant progressivement le
principe actif, dans le but de remédier aux défauts des formes
galéniques classiques, et plus particulièrement, éviter la
nécessité d'administrations répétées
(V. Michel, 1996). Par ailleurs, plus récemment, il y a
eu un grand intérêt pour le développement de vecteurs de
principes actifs qui utilisent des nanoparticules, des microparticules
composés de polymères biodégradables. Ces matrices
polymères modifient la libération, la pharmacocinétique et
la distribution de principes actifs dans l'organisme. En effet, ces
progrès vont sans doute changer le développement des
médicaments. Au lieu de chercher de nouvelles molécules, il sera
possible de modifier les propriétés pharmacodynamiques des
médicaments existants en manipulant différents systèmes de
vecteurs (i.e. faire du neuf avec du vieux en utilisant les nouvelles
technologies des vecteurs de médicaments) (O . Hung,
2006). La littérature fournit des exemples variés
d'application intenses des polymères biodégradables à base
de PLA dans le domaine de vectorisation de principes actifs, notamment des
stéroïdes, agents anticancéreux, peptides, protéines,
antibiotiques, anesthésiques et vaccins.
L'utilisation des principes actifs anti-inflammatoires non
stéroïdiens est souvent limitée par la
nécessité de véhiculer le principe actif vers le site
spécifique de l'organe ou tissu ciblé. L'utilisation des
anti-inflammatoires non stéroïdiens (AINS) est aussi limitée
par leurs effets secondaires irritants dans la muqueuse gastro-intestinale et
par leurs faibles solubilités dans l'eau. Cependant, ces
problèmes peuvent être surmontés par la préparation
des systèmes polymère/principe actif à partir des liaisons
hydrolysables (M. Babazadeh, 2006). L'ibuprofène (IB)
est l'un des meilleurs principes actifs de la famille des AINS valable pour le
traitement du rhumatisme articulaire, ostéoarthrite et pour le
soulagement des douleurs. L'ibuprofène est bien adapté pour le
traitement de la fièvre, la douleur, la migraine, la
dysménorrhée et les douleurs arthritiques chroniques. En plus,
l'ibuprofène est rapidement absorbé dans le corps et
présente une demi-vie courte (~ 2 h) ce qui nécessite des
administrations répétées. Cependant,
le ralentissement de la vitesse de libération de l'IB
à partir de la formulation, en utilisant des
formes galéniques
à libération prolongées, pourraient réduire la
fréquence d'administration du
principe actif et par conséquent
ses effets secondaires seront réduites ou éliminés
(C. De
Brabander, 2004).
Récemment, la littérature nous a montrée
que des matrices polymères biodégradables ont été
employées pour prolonger les vitesses de libération de
l'ibuprofène. Ces systèmes ont montré d'excellentes
propriétés de vectorisation de médicaments. Par exemple,
Juliana Baidone et al. (J. Bidone, 2009), ont réussi
à avoir des vitesses de libération prolongées de
l'ibuprofène en utilisant des matrices polymères blindes de type
P(3HB):mPEG-PLA. Ces vitesses de libération sont désirables pour
l'administration intra-articulaire. T. Phromsopha et Y. Baimark (T.
Phromsopha, 2009), ont montré que des films à base de
polymères biodégradables tels les films de méthoxy
poly(éthylène glycol)-b-poly(D,L-lactide) (MPEGb-PDLL) sont
très intéressants pour la vectorisation de principes actifs
hydrophobes tel l'ibuprofène.
Les interactions entre la matrice polymère et le
principe actif présentent un paramètre critique qui influe sur
les systèmes de vectorisation de principes actifs. Ces interactions
jouent un rôle significatif sur les profils de libération du
principe actif car ces interactions peuvent induire des changements dans le
système polymère/principe actif qui peuvent être utiles
pour un processus de libération contrôlé du principe actif
(C. S. Proikakis, 2006).
Pour notre travail, nous avons envisagé de
synthétiser un polymère biodégradable qui est le
poly(D,L-acide lactique) et étudié des éventuelle
interactions avec l'ibuprofène, en utilisant diverses méthodes de
préparations des mélanges. L'objectif de cette étude est
d'étudié l'effet de certains paramètres tels la
concentration et la masse moléculaire du PDLLA, le pH du milieu de
dissolution et la méthode de préparation du mélange
IB/PDLLA sur la cinétique de libération de
l'ibuprofène.
Le premier chapitre de ce manuscrit « étude
bibliographique » est divisé en deux parties. Dans la
première partie, nous nous attacherons d'abord à une étude
bibliographique sur le poly(acide lactique) (PLA) ou nous parlerons sur sa
structure chimique, ses méthodes de synthèse, ses
propriétés ainsi que sa production industrielle. Dans la seconde
partie, nous allons parler des caractéristiques physico-chimiques de
l'ibuprofène, ses caractéristiques, sa pharmacocinétique,
son utilisation thérapeutique, ses effets indésirables et enfin
sur l'utilisation de l'ibuprofène dans le domaine de la vectorisation de
médicaments. Dans le deuxième chapitre, nous nous attacherons
à définir le concept général de vectorisation de
principes actifs et le rôle des polymères dans la
libération de principes actifs. Ensuite, l'accent sera porté sur
la présentation de quelques exemples d'application du poly(acide
lactique) dans le domaine de la vectorisation de principes actifs.
Le troisième chapitre de ce manuscrit « partie
expérimentale » traitera, dans la première partie, de la
synthèse du poly(D,L-acide lactique) par polycondensation
azéotropique du D,Lacide lactique ainsi que la détermination des
masses viscosimétriques, en utilisant un viscosimètre de type
Ubbelhode. La caractérisation des PDLLAs synthétisés ainsi
que l'ibuprofène offert par le groupe Saidal par différentes
techniques d'analyses (IRTF, DSC, DRX, ATG-ATD) sera aussi traité dans
cette partie. La deuxième partie de ce chapitre
traitera, d'abord, des différentes méthodes de
préparation des mélanges IB/PDLLA (mélange physique,
mélange par fusion à chaud et mélange par
évaporation de solvant). Ensuite, nous passerons à la
caractérisation des différents mélanges par les
différentes techniques de caractérisation (IRTF, DRX, MEB).
Enfin, nous terminerons par une étude de la cinétique de
dissolution de l'ibuprofène à partir des formulations
préparées, dans différents milieux physiologiques. Le
quatrième chapitre sera consacré à la discussion des
résultats obtenus.
SECTION BIBLIOGRAPHIQUE
Chapitre I. Rappels bibliographiques sur le
poly(acide lactique) et l'ibuprofène
Chapitre I. Rappels bibliographiques sur le
poly(acide lactique) et
l'ibuprofène
Partie A
A-I. Poly(acide lactique) (PLA)
Parmi les polyesters aliphatiques biodégradables, le
poly(acide lactique) a reçu l'attention des scientifiques à cause
de son rôle très important dans plusieurs domaines. En effet, le
monomère de base de ce polymère (i.e. acide lactique) peut
être produit en grande quantité par fermentation à partir
des ressources renouvelables (matériaux féculents et sucres). Le
poly(acide lactique) est un thermoplastique qui présente de bonnes
propriétés mécaniques (ex. résistance, modulation),
qui peut être traité par des techniques conventionnelles (moulage
par injection, moulage par soufflage, thermoformage et extrusion). Pour une
production à grande échelle, le PLA doit présenter une
stabilité thermique adéquate pour prévenir la
dégradation et aussi lui permettre de maintenir sa masse
moléculaire et ses propriétés intactes (A. P.
Gupta, 2007).
Le poly(acide lactique) est très utilisé dans le
domaine médical pour ces diverses propriétés à
savoir biorésorbabilité, biodégradabilité,
biocompatibilité et de bonnes propriétés
mécaniques. Grâce à ses propriétés, un grand
nombre d'études ont été effectuées sur le PLA et
ses copolymères dans le domaine biomédical. Il a
été très utilisé dans les systèmes à
libération prolongée, suture chirurgical, chirurgie
orthopédique et technologie tissulaire (Z. Zhong-cheng,
2005).
A-I.1. Le monomère de base de poly(acide
lactique)
L'unité de base constitutive des chaines de PLA est
l'acide lactique. Ce dernier a été isolé pour la
première fois en 1780 à partir du lait coagulé par le
chimiste suédois SCHEELE, et il a été produit
commercialement en 1881 (D. Garlotta, 2001 ; E. Rudnik, 2008).
Le monomère « acide lactique » peut être
synthétisé par deux méthodes chimique et biologique. En
effet, la voie pétrochimique était la méthode la plus
répondue jusqu'à 1990. L'acide lactique préparé par
cette méthode est un mélange racémique des
énantiomères D et L.
Aujourd'hui, la méthode biologique est
généralement la plus utilisée puisque la majorité
de l'acide lactique mondial est produit par fermentation bactérienne.
Cette dernière est basée sur la fermentation de l'amidon et
d'autres polysaccharides, lesquels sont disponibles à partir du
blé, sucre de betterave, sucre de la cane, pomme de terre et d'autres
formes de biomasses. Durant le processus de la fermentation, des
paramètres comme pH, température, pression et dans certains cas
l'agitation sont hermétiquement contrôlés dans le but
d'avoir un rendement maximum en produit pur (A. P. Gupta,
2007). L'acide lactique ainsi produit est exclusivement de type
isomère L (> 99,5 %) (B. Gupta, 2007).
Le monomère utilisé pour la production de
poly(acide lactique) est obtenu à partir des récoltes
annuellement renouvelables. En effet, l'énergie solaire provoque la
photosynthèse dans les cellules végétales (plantes
vertes), ensuite le dioxyde de carbone (CO2) et la vapeur d'eau (H2O) de
l'atmosphère sont transformés en amidon. L'amidon est extrait
facilement de
la plante est transformé en sucre fermentable (ex.
glucose) par hydrolyse enzymatique. Enfin, le sucre naturel obtenu est
transformé vers acide lactique par fermentation (R. S.
Blackburn, 2005) (figure 1).

CO2 + H2O Photosynthèse
H
*
O
H
HO
O
H
OHH
O
HO
H
*
n
Amidon

+ H2O
Hydrolyse enzymatique
O
H H
H
H
H
HO
H
O
Fermentation
HO
HO
H
H
CH3
HO
OH
O
Glucose
Figure 1. Production de l'acide lactique
à partir des ressources renouvelables (R. S. Blackburn,
2005).
L'acide lactique (ou acide 2-hydroxypropanoïque) est une
molécule chirale simple quiexiste sous deux formes
énantiomères optiquement actives (figure 2). La forme
dextrogyre,
appelée L(+)-acide lactique ou (S)-acide lactique, et
la forme lévogyre, appelée D(-)-acide lactique ou (R)-acide
lactique. La forme D,L- ou méso est optiquement inactive, c'est le
mélange équimolaire (racémique) des deux
énantiomères D et L. Acide lactique est produit dans les muscles
des mammifères durant le processus de glycogénèse (i. e.
formation du glucose à partir de glycogène) et il est
impliqué dans le cycle de Krebs à partir de l'acide pyruvique et
de l'acétyl-CoA (B. Gupta, 2007 ; K. J. Jem, 2010).

acide D-lactique acide L-lactique
Figure 2. Différents isomères
optiques de l'acide lactique (A. P. Gupta, 2007).
A-I.2. Méthodes de synthèse de poly(acide
lactique)
La synthèse de poly(acide lactique) s'effectue selon trois
méthodes de polymérisation différentes, comme le montre la
figure 3.
O
O
HO
OH
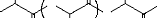
CH3
O
n
CH3
O CH3 O
Polymère de faible masse
moléculaire
Mw = 2000 à 10,000


O
CH3
O
CH3
O
n
CH3
O
O
Agents d'accouplement des chaînes
HO
OH

CH3
O
OH
Acide lactique
HO
- H2O Condensation

CH3
O
HO
O
O
n
CH3
CH3
O
O

Polymère de masse moléculaire
faible
Mw = 1000 à 5,000
PLA de masse moléculaire
élevée
Mw > 100,000

Polymérisation par ouverture de cycle
OH
O O CH3
H3C O
O
Lactide
Figure 3. Méthodes de synthèse
du PLA (D. Garlotta, 2001).
A-I.2.1. Polymérisation par condensation directe en
absence de catalyseur
La première méthode est dite directe où
l'acide lactique est polymérisé par condensation pour donner un
polymère de faible masse moléculaire, de nature vitreux et
cassant. Ce type de polymère est, dans la majorité des cas,
inutilisable à moins que si des agents de couplage externes sont
utilisés pour augmenter sa masse moléculaire. La masse
moléculaire faible de ce type de polymère est à l'origine
de plusieurs raisons, on cite ce qui suit : présence d'eau,
impuretés, la faible concentration des groupements réactifs
terminaux et un équilibre de réaction engendrant le cycle du
lactide à six chaînons. La deuxième méthode est dite
indirecte, il s'agit de la polymérisation par ouverture de cycle de
lactide. Cette méthode nous permet d'avoir un polymère pur et de
masse moléculaire élevée (Mw > 100.000)
(D. Garlotta, 2001). La polymérisation de l'acide
lactique exige des monomères de pureté élevée, car
les
impuretés présentent une barrière devant
le développement de la réaction et réduisent par
conséquent la qualité de polymère attendu. Des groupements
fonctionnels comme hydroxyle, carboxyle et eau, etc. peuvent être
considérés comme des impuretés. En effet,
l'impureté hydroxyle influe à travers des réactions de
formation de l'initiateur, transfert de chaînes, et la
transestérification, ce qui provoque l'augmentation de la vitesse de la
réaction de polymérisation et la diminution de la masse
moléculaire de polymère obtenu. Les impuretés
carboxyliques provoquent l'inhibition de la polymérisation en formant un
complexe avec le catalyseur ce qui diminue la vitesse de la réaction,
mais sans montrer aucun effet considérable sur la masse
moléculaire de polymère final. Généralement, il
existe quatre méthodes de synthèse de poly(acide lactique)
(A. P. Gupta, 2007).
A-I.2.2. Polymérisation par condensation directe en
présence de catalyseur
Le monomère acide lactique est polymérisé
en présence d'un catalyseur sous une pression réduite (figure 4).
Le polymère obtenu a une masse moléculaire faible, car il est
difficile d'enlever l'eau complètement à partir du mélange
réactionnel de viscosité élevée ; par
conséquent, le polymère obtenu possède une masse
moléculaire de quelques dizains de milliers. La faible masse
moléculaire est l'inconvénient principal de la
polymérisation par polycondensation directe, ce qui a limité son
utilisation à moins que si le domaine d'application de ce
polymère exige qu'il soit de faible masse moléculaire. Cependant,
un polymère de haut poids moléculaire peut être obtenu au
moyen des agents d'accouplement de chaînes. Ces agents permettent de
relier une chaîne de polymère de faible masse avec une autre
chaîne de polymère de masse élevée. Les agents
d'accouplement de chaînes réagissent avec les deux groupements
terminaux (-OH et -COOH) (A. P. Gupta, 2007).
H2O

O
H
OH
CH3
HO
*
CH3
O
H
O *
Acide lactique Poly(acide lactique)
Figure 4. Synthèse de PLA par
polycondensation direct (D. E. Henton, 2005).
Avec l'utilisation d'un co-monomère
bi/multifonctionnels, les chaînes de PLA peuvent être
modifiées. En effet, les chaînes de PLA terminées par un
hydroxyle sont obtenues par polymérisation de l'acide lactique en
présence d'une petite quantité d'un composé
bi/multifonctionnel, citant par exemple : 2-butène 1,4-diol,
glycérol ou 1,4-butanediol, lesquels mènent a un groupement
hydroxyle préféré. Ce même concept peut être
utilisé pour la synthèse d'un PLA terminé par un
carboxyle, l'utilisation d'un acide carboxylique bi/multifonctionnels (acide
maléique, succinique, adipique,...) nous donne un polymère avec
un groupement carboxylique terminale. Le PLA peut aussi réagir avec un
anhydride acide (acide maléique ou succinique) pour convertir un
groupement hydroxyle vers un groupement carboxyle terminal (A. P.
Gupta, 2007 ; D. Garlotta, 2001).
A-I.2.2.1. Polymérisation par condensation
azéotropique
Dans cette approche, le problème d'élimination
de la vapeur d'eau est surmonté. En effet, un équilibre entre
monomère et polymère est provoqué, l'acide lactique est
donc polycondensé directement en donnant un polymère de masse
moléculaire élevée. Ajioka et al. (M. Ajioka, 1995
; M. Ajioka, 1998) ont synthétisé un polymère
(PLA) de haut poids moléculaire par une étape de
polymérisation par condensation azéotropique de l'acide lactique
en utilisant un solvant azéotropique approprié. Cette technique
de polymérisation réclame l'utilisation d'un catalyseur
très actif et d'un solvant peut volatil. L'eau comme sous produit est
extrait azéotropiquement, par contre le solvant est
entraîné sous reflux au sein de la réaction. Cette
technique de polymérisation permettre le choix de la température
réactionnel de telle sorte qu'elle soit inférieure a la
température de fusion de polymère, et ainsi la
dépolymérisation et la racémisation sont
empêchées d'une manière efficace durant le processus de
polymérisation. Il a été montré qu'au moyen de
cette méthode, un polymère (PLA) ultrapur et de masse
moléculaire d'environ 300 000 g/mol peut être obtenu (A.
P. Gupta, 2007).
A-I.2.2.2. Polymérisation à l'état
solide
Ce processus implique le chauffage d'un
prépolymère solide et semi-cristallin (de masse
moléculaire relativement faible) sous forme de poudre, granules,
morceaux ou fibres jusqu'a une température inférieure a sa
température de fusion, suivi par une extraction des sousproduits a
partir de la surface du matériau soit sous une pression réduite
(volatilisation) ou avec un vecteur (ex. gaz inerte). Ce dernier serve a
extraire le condensat et d'empêcher l'oxydation de polymère
(figure 5). Ces réactions s'effectuent essentiellement dans la
région amorphe de polymère, où les groupements terminaux
réactifs résident. Par conséquent, la température
réactionnel doit être inférieure a la température de
transition vitreuse (pour faciliter la mobilité des groupements
terminaux et d'augmenter leur activité), et aussi inférieure a la
température de fusion de polymère. En plus, la réaction a
l'état solide commence a des températures plus faibles
comparées a celles a l'état fondu ou soluble (elles peuvent
variées de 5 a 15 °C). Mais, pour faciliter la croissance des
chaînes de polymère, la température de
polymérisation des monomères doit être suffisamment
élevée mais pas aussi élevée qu'elle mène a
des réactions secondaires (par exemple cyclisation). Avec cette
technique de polymérisation, de très hauts poids
moléculaires peuvent être atteints, mais a la différence de
la technique de polymérisation a l'état soluble, pour aboutir a
de tels poids il est important de laisser la réaction se déroule
sur un intervalle de temps assez large (A. P. Gupta, 2007).
Etat fondu Etat solide

HO
CH3
O
OH
Catalyseurs/vide
150-180°C
H
O
CH3
O
a
OH
au-dessous de Tf
Vide ou gaz inert
H
O
CH3
O
n
OH
Acide lactique Oligomère PLA de masse moléculaire
élevée
a = 8 - 15
Figure 5. Polymérisation a
l'état solide (A. P. Gupta, 2007).
Cette technique de polymérisation présente
plusieurs avantages, des températures opérationnelles faibles
permettent de contrôler les réactions secondaires
(dégradation thermique, dégradation oxydative, dégradation
hydrolytique) et cela réduit la décoloration et la
dégradation des produits. Enfin, la polymérisation à
l'état solide ne présente aucune pollution environnementale car
elle n'exige pas des solvants organiques (A. P. Gupta,
2007).
A-I.2.3. Polymérisation par ouverture de cycle
Elle a été mise en oeuvre pour la
première fois par CAROTHERS en 1932. Comme il est possible de
contrôler les propriétés du polymère obtenu de
plusieurs manières, cette méthode trouve un champ d'application
très large. En effet, cette technique de polymérisation est
employée pour la synthèse d'un polymère de haut poids
moléculaire. Récemment, la société Mitsui Toatsu
Chemicals a synthétisé un PLA avec une masse moléculaire
supérieure à 300,000 g/mol en procédant de façon
suivante, l'acide lactique et le catalyseur ont été
azéotropiquement déshydratés sous reflux, à chaud,
en présence d'un solvant aprotique et sous une pression réduite
(A. P. Gupta, 2007 ; D. Garlotta, 2001 ; B. Gupta, 2007).
Le principe de cette méthode est basé sur la
polymérisation d'un dimère cyclique à six chainon
appelé « lactide ». Ce dernier est préparé
à partir de craquage d'un oligomère PLA de faible poids
moléculaire à haute température et sous une pression
réduite en présence d'un catalyseur (figure 6).
O

O
O
O
CH3
H3C
O
*
n
CH3
*
O
H
H
HO
OH
CH3
Polymérisation
O
Acide lactique Lactide Polylactide
Figure 6. Synthèse de PLA par
polymérisation par ouverture de cycle (A. P. Gupta,
2007).
Le cycle lactide (i.e. 3,6-diméthyl 1,4-dioxane
2,5-dione) présente, à la différence de monomère
acide lactique, trois stéréoisomères optiques : DD-, LL-,
et DL-lactide comme le montre la figure 7 (A. P. Gupta, 2007 ; B.
Gupta, 2007).

O
O
O
O
O
CH3
O
O
H3C
O
CH3
CH3
O
O
H3C
O
O
H3C
DD-lactide DL-lactide LL-lactide
Figure 7. Les différents
isomères du lactide (A. P. Gupta, 2007 ; B. Gupta,
2007).
Le monomère lactide doit être purifié
avant polymérisation, car il contient des impuretés comme eau,
acide lactique et une oligomère. En effet, ces impuretés
interférent avec la réaction de polymérisation ce qui
donne comme résultat la formation d'un polymère de masse
moléculaire faible avec un degré de racémisation
élevé.
Selon les mécanismes et les types d'amorceurs
utilisés, la réaction de polymérisation par ouverture de
cycle peut être classifiée en trois types de réactions les
plus répondues (A. P.
Gupta, 2007).
A-I.2.3.1. Polymérisation anionique
L'étape d'amorçage de ce type de
polymérisation débute lors de l'attaque de l'initiateur anionique
(nucléophile) (par exemple alkoxyde des métaux alcalins) sur le
groupement carbonyle de lactide, résultant la rupture de la liaison
entre l'atome de carbone du carbonyle et l'atome d'oxygène endocyclique.
Cette dernière devient un site négatif (formation d'un groupement
alkoxyde terminal), l'attaque de ce site négatif sur le carbonyle d'un
autre cycle de lactide constitue l'étape de propagation, le
mécanisme de cette méthode est schématisé dans la
figure 8 (A. P. Gupta, 2007 ; D. Garlotta, 2001),. Cependant,
l'initiateur nucléophile est suffisamment basique qu'il peut provoquer
la déportonation du monomère, ce qui mène à la
racémisation. Dans ce cas, des catalyseurs très actifs et
à haute température provoquent des réactions inter et
intra-moléculaires ainsi que d'autres réactions secondaires, ce
qui limite la propagation des chaînes de polymère. Par
conséquent, il est très difficile d'obtenir un polymère de
haut poids moléculaire avec cette méthode (A. P. Gupta,
2007).

R
O
Figure 8. Polymérisation par ouverture de
cycle anionique (A. P. Gupta, 2007).
CH3
Lactide
CH3
O
O
O:M
O
n
O
CH3
CH3
O
R
O
O
O:M
R O : M
+
O
O O
H3C
CH3
O
A-I.2.3.2. Polymérisation cationique
L'étape d'amorçage de cette technique de
polymérisation débute lorsque l'atome d'oxygène
exocyclique de l'un des groupements carbonyles du lactide est soit
alkylé ou protoné par l'initiateur, en donnant naissance à
la liaison O-CH chargée positivement. Une attaque nucléophilique
par un deuxième monomère provoque la rupture da cette liaison
pour créer un autre site électrophile. L'addition continu du
monomère assure, dans l'étape de propagation, une succession
d'attaques nucléophiles jusqu'à ce que la polymérisation
se
termine par un nucléophile monofonctionnel comme l'eau,
le mécanisme de cette technique de polymérisation est
schématisé dans la figure 9. Les initiateurs utilisés dans
la polymérisation cationique peuvent être des carbanions donneurs
et quelques acides forts comme triéthyloxoniumtetrafluoroborate,
trifluorure de bore et acide trifluoroacétique.
Dans la polymérisation cationique, des
températures élevées provoquent une racémisation
car le monomère attaque sur un centre chiral de la chaîne de
propagation. Cependant, la racémisation peut être minimisée
à des températures >50°C, mais à ce niveau la
vitesse de la réaction devient faible et la masse moléculaire de
polymère diminue (A. P. Gupta, 2007).

F3CSO3CH3 +
H3C
O
O
O
CH3
H3C
OCH3
O
O
CH3
O
O
+ F3C SO3

H3C
O
H3C
O
CH3
O
CH3
O
O
O
O
O
O
O
O
CH3
CH3
CH3
OCH3
O
O
OCH3
O H3C
Figure 9. Polymérisation par ouverture de
cycle cationique (A. P. Gupta, 2007 ; D. Garlotta, 2001).
A-I.2.3.3. Mécanisme de coordination-insertion
C'est la méthode la plus étudiée pour la
synthèse d'un polymère de haut poids moléculaire. Les
catalyseurs utilisés dans cette méthode contient des orbitales
«p» et «d» libres d'énergie avantageux (par exemple
alkoxydes de Mg, Zn, Zr, Ti, Sn), ces catalyseurs possèdent une liaison
covalente entre l'atome métallique et l'atome d'oxygène, et ainsi
se comportent comme des acides de Lewis.
Le mécanisme de coordination-insertion peut être
expliqué comme suit, d'abord, le mécanisme commence par la
coordination de l'atome d'oxygène exocyclique du lactide avec l'atome
métallique de l'initiateur. Cette coordination temporaire provoque
l'augmentation du caractère nucléophile de la partie alkoxide de
l'initiateur ainsi que le caractère électrophile de groupement
carbonyle de lactide. Ensuite, la liaison acyl-oxygène (entre le
groupement carbonyle et l'atome d'oxygène exocyclique) du lactide est
rompue et la chaîne de lactide produite est insérée sur la
liaison acyl-oxygène de l'initiateur. La polymérisation se
poursuivre pendant que les molécules de lactide sont ouvertes et
insérées sur la liaison entre l'atome métallique et
l'atome d'oxygène adjacente, tandis que l'autre extrémité
i.e. l'alkoxide terminale de l'initiateur devient une extrémité
d'une chaîne inactif, la figure 10 présente le mécanisme de
coordination-insertion. Avec cette méthode de synthèse, des
polymères de haut poids moléculaire peuvent être obtenus
(A. P. Gupta, 2007).
En conclusion, l'utilisation de catalyseurs à base de
métaux lourds (ex. oxydes de Zn et Sn, chlorures de Zn et Sn, ou octoate
d'étain) dans le processus de polymérisation par ouverture
de cycle a contribué à la contamination des
polymères obtenus et par conséquent leurs application dans les
domaines de l'emballage et les systèmes médicales sera
limitée. Pour remédier à se problème, certains
chercheurs ont proposés certains solutions, à savoir le choix de
catalyseurs non toxiques, des traitements spécifiques pour le produit
obtenu ou utilisation de nouvelles voix de synthèse plus propres. L'une
des alternatives utilisées pour éliminer ces problèmes et
la polymérisation enzymatique. Cette dernière est avantageux car
c'est une méthode environnementale (i. e. propre), elle peut s'effectuer
sous des conditions douces et enfin le processus de polymérisation peut
être contrôlé (Y. Cheng, 2009).

O
O
O
OR
Al
O OR
O
O
CH3
O
Al(OR)3
O
H3C
RO
CH3
H3C
O
O
O
O
O
H3C
OR
O
RO RO
Al
O
O
H3C
CH3
CH3

RO
CH3
OR
Al
OR
n
Lactide
O
O
CH3 O
O
CH3 RO OR
O
O
O
Al
O
m
H3C
O
CH3
CH3
H3O
H
RO
O n
RO
O
O
Figure 10. Mécanisme de
coordination-insertion (A. P. Gupta, 2007).
A-I.3. Catalyseurs utilisés dans la synthèse
de PLA
En général, les catalyseurs utilisés
consistent à des poudres métalliques, acides et bases de Lewis,
composés organométalliques et de différents sels
métalliques. Les catalyseurs organométalliques et en particulier
les alkyles métalliques, les halogénures métalliques, les
oxydes, les carboxylates et les alkoxydes sont très efficaces dans la
synthèse d'un polymère de haut poids moléculaire. Un grand
nombre de catalyseurs ont été étudiés pour la
polymérisation de lactide pour de divers applications, citant entre
autres fer (Fe), Sn(Oct)2, SnCl4, SnCl2, Sn(C6H6)4, lactate de zinc
[(n-C4H9O2)AlO]2Zn. Des composés organiques comme les éthers
couronne sont très efficaces pour la synthèse de PLA de
pureté optique élevée et de haut poids moléculaire.
De hauts poids moléculaires sont aussi obtenus lorsqu'un initiateur de
type chlorure de lithium est utilisé avec l'éthylène
glycol et a-D-
glucopyranoside. Certains catalyseurs comme
oxyéthylméthacrylatetrialkoxyde d'aluminium, alkoxyde
d'étain cyclique, lithium et magnésium butyliques,
diisopropylamide de lithium, bis(triméthyltriazacyclo hexane)
peraseodymiumtriflate, yttrium (Y), et tris(isopropoxyéthoxyde) de
yttrium sont trouvés très actifs dans la polymérisation de
D,L-lactide dans une solution de dichloro-méthane. Des composés
à base d'étain, en particulier acide Sn(II)
bis-2-éthylhéxanoïque (Sn(Oct)2) (figure 11), sont
préférés pour la polymérisation de lactide à
cause de leur solubilité dans le lactide fondu ainsi que dans les
solvants organiques, activité catalytique élevée, et
stabilité en cas de stockage. Le mécanisme prévu est aussi
de type coordination-insertion comme le montre figure 12. Polymère de
haut poids moléculaire, vitesse réactionnelle
élevée, et faible degré de racémisation de
polymère sont observés dans la réaction de
polymérisation de lactide catalysée par (Sn(Oct)2). Des
conditions typiques sont à considérés dans ce type de
polymérisation à savoir, température variant de 180
à 210 °C, des concentrations en (Sn(Oct)2) entre 100-1000 ppm, et
un temps de 2 à 5 heures pour atteindre environ 95 % de rendement
(A. P. Gupta, 2007 ; B. Gupta, 2007).

Sn
Figure 11. Octoate d'étain (A.
P. Gupta, 2007).
De nombreux composés de terre rare ont été
aussi étudiés dans le cas de la polymérisation par
ouverture de cycle, citant triphénylyttrium,
triphénylnéodymium et triphénylsamarium. Iia
été montré que l'utilisation du catalyseur
triphénylyttrium abouti à des polymères de hauts
poids moléculaires pour un rapport
monomère/initiateur faible. Un mécanisme de type
coordination-déprotonation-insertion a été
suggéré dans le cas de 2-méthylphénylsamarium et un
mécanisme de type rupture de la liaison acyl-oxygène a
été suggéré dans le cas de lanthanide
(2,4,6-triméthylphénolate) (A. P. Gupta,
2007).
H3C

CH3
CH3
CH3
ROH
Sn(Oct)2
H3C
H
H3C
H3C
H
O
R
Sn
O
O
O
O
O
O
O
O
(Oct)2
O
O
O
O O Sn R (Oct)2

O
CH3
H
O
CH3 O Sn(Oct)2
O
O O
CH3
H
Sn(Oct)2
H3C
O
O
RO
Figure 12. Polymérisation de lactide
par le mécanisme coordination-insertion catalysé par l'octoate
d'étain (B.
Gupta, 2007 ; D. E. Henton, 2005).
A-I.4. Propriétés de poly(acide lactique)
A-I.4.1. Propriétés thermiques
Comme tout polymère thermoplastique, le poly(acide
lactique) présente une température de transition vitreuse
(Tg ~58 °C) relativement élevée. À des
températures supérieures à Tg, le PLA est
caoutchouteux, et dans le cas contraire (i.e. T < Tg) il devient
vitreux et cassant. Comparé aux autres polymères thermoplastiques
cristallins et semi-cristallins, le PLA présente une température
de fusion relativement faible. Chacune des températures Tg et
Tf est fonction de la masse moléculaire et de la pureté optique.
La température de transition vitreuse augmente avec la masse
moléculaire et avec la cristallinité du PLA. En effet, le PLA
constitué d'une grande quantité d'isomère L-lactide
possède une température de transition vitreuse supérieure
à celle de PLA riche en isomère D-lactide. Pour un PLA
stéréochimiquement pur (soit D ou L), la température de
fusion est pratiquement aux environs de 180°C et une enthalpie de 40-50
J/g. La présence de lactide (méso) dans la structure de PLA peut
faire diminuer Tf d'environ 50°C. Cette diminution dépend de la
quantité de D-lactide incorporée dans le polymère. Cette
baisse de Tf présente plusieurs implications importantes, à
savoir réduction de la dégradation thermique et hydrolytique et
affaiblissement de la formation de lactide. D'autre propriétés du
PLA sont données dans le tableau 1 suivant (L.-T. Lim,
2008)
Tableau 1. Propriétés thermiques
typiques de poly(acide lactique) (J.-F. Zhang, 2005).
|
Propriétés
|
Unités
|
Conditions
|
Valeurs
|
|
Chaleur de fusion, ÄHf
|
KJmol-1
|
L-PLA de cristallinité complète
|
146
|
|
|
Fibre de L-PLA
|
|
|
|
Après extrusion
|
2.5
|
|
|
Après étirage à chaud
|
6.4
|
|
Capacité thermique, Cp
|
JK-1g-1
|
L-PLA de :
|
|
|
|
Mv = 5300
|
0,60
|
|
|
Mv = (0,2-6,91) õ 105
|
0,54
|
|
Température de transition vitreuse, Tg
|
K
|
L-PLA de différents poids moléculaires
|
326-337
|
|
|
D,L-PLA de différents poids moléculaires
|
323-330
|
|
Température de fusion, Tf
|
K
|
D-PLA moulé à chaud
|
444,4
|
|
|
Mv = 1000
|
|
|
|
L-PLA de différents poids moléculaires
|
418-459
|
|
Température de décomposition,
Td
|
K
|
L-PLA de Mw = (0,5-3) õ 105 D, L-PLA
de
|
508-528
|
|
|
Mw = (0,21-5,5) õ 105
|
528
|
A-I.4.2. Propriétés mécaniques
Les propriétés physiques et mécaniques de
PLA dépendent de divers facteurs, rapport L/D, masse moléculaire,
orientation des chaînes, et méthodes de préparation.
L'inconvénient le plus important de PLA utilisé comme plastique
est sa faible flexibilité. Le poly(acide lactique) est un
matériau rigide, cassant, et probablement déformable à des
températures supérieures à Tg. Donc, il est
préférable d'améliorer les propriétés
mécaniques de PLA pour élargir son domaine d'application. En
effet, les propriétés mécaniques peuvent être
améliorées par le contrôle du rapport L/D ou par
l'utilisation d'un catalyseur spécifique lors de la
polymérisation de PLA. Le poly(acide lactique) faiblement
enchevêtré et réticulé peut être
polymérisé à une température inférieure
à sa température de fusion, et le polymère ainsi obtenu
montre une grande résistance mécanique (jusqu'à 805
MPa).
Le degré de cristallinité d'un polymère
dépend de plusieurs facteurs, citant la masse moléculaire,
traitement thermique et le temps de traitement thermique. Poly(L-acide
lactique) est cristallin par contre le poly(D, L-acide lactique) est amorphe.
À cause de cette différence de cristallinité, PLLA
présente de meilleures propriétés mécaniques que
PDLLA sachant qu'ils ont la même masse moléculaire. En plus, PLLA
traité à chaud possède de meilleures
propriétés mécaniques que celui non traité à
cause de l'augmentation de la cristallinité sous l'effet de chauffage.
PLA lentement cristallisé devient très résistant, cela
montre que la présence des domaines cristallins à un effet
positif sur la ductilité. PLA de masse moléculaire
élevée présente une résistance mécanique
importante, par exemple, l'augmentation de la masse moléculaire de PLLA
de 23 à 67 KDa, la résistance à la flexion sera
augmentée de 64 à 106 MPa mais la résistance à
l'étirement reste la même 59 MPa. L'augmentation de la masse
moléculaire de PDLLA de 47,5 à 114 KDa, la résistance
à la flexion et la résistance à l'étirement seront
augmentées de 49 à 53 MPa et de 84 à 88 MPa,
respectivement (A. P. Gupta,
2007). Le poly(acide lactique) amorphe d'un
réseau orienté possède une résistance à
l'étirement aux environ de 460 MPa. L'auto-renforcement et
l'étirement à chaud améliore aussi les
propriétés mécaniques de PLA. En effet, ce ci peut
être réalisé par l'alignement des molécules de
polymère pour avoir un degré d'orientation élevé
des chaînes, c'est la transformation d'une structure sphérique
vers une structure fibreuse. Le tableau 2 suivant résume les
propriétés mécaniques typiques du PLA (J.-F.
Zhang, 2005).
Tableau 2. Propriétés
mécaniques typiques de polyacide lactique.
|
Propriétés
|
Unités
|
Conditions
|
Valeurs
|
|
Limite élastique à la
traction
|
|
Film ou disque de PLLA, Mw= (0,5-3) x
10-1
|
28-50
|
|
MPa
|
Fibres PLLA
|
Jusqu'à 870
|
|
|
Film ou disque de PDLLA, Mw= (1,07-5,5) x
105
|
29-35
|
|
Module d'élasticité
|
|
Film ou disque de PLLA, Mw= (0,5-3) x
10-1
|
1200-3000
|
|
MPa
|
Fibres PLLA
|
Jusqu'à 9200
|
|
|
Film ou disque de PDLLA, Mw= (1,07-5,5) x
105
|
1900-2400
|
|
Module de stockage
à la flexion
|
MPa
|
Film ou disque de PLLA, Mw= (0,5-3) x
10-1
|
1400-3250
|
|
|
Film ou disque de PDLLA, Mw= (1,07-5,5) x
105
|
1950-2350
|
|
Allongement à
l'extrémité
élastique
|
|
Film ou disque de PLLA, Mw= (0,5-3) x
10-1
|
3,7-1,8
|
|
%
|
Film ou disque de PDLLA, Mw= (1,07-5,5) x
105
|
4,0-2,5
|
|
|
Film ou disque de PLLA, Mw= (0,5-3) x
10-1
|
6,0-2,0
|
|
Allongement à la
|
|
Fibres PLLA dans le toluène
|
12-26
|
|
rupture
|
%
|
Fibres PLLA Mw= 1,8 x 105
|
25
|
|
|
Film ou disque de PDLLA, Mw= (1,07-5,5) x
105
|
6,0-5,0
|
|
Résistance aux
cisaillements
|
MPa
|
PLLA
|
54,5
|
|
Module de
cisaillement
|
MPa
|
Mono-filament de PLLA
|
1210-1430
|
|
Résistance à la
flexion
|
MPa
|
PLLA
|
132
|
|
Module de flexion
|
MPa
|
PLLA
|
2800
|
A-I.5. Vieillissement et biodégradation de
poly(acide lactique)
A-I.5.1. Vieillissement physique de poly(acide
lactique)
Le vieillissement physique, comme phénomène
général, est caractéristique de l'état vitreux de
tout matériau et il est thermoréversible. Il peut être
éliminé par un simple chauffage du matériau. Le
vieillissement est peut être attribué à la relaxation des
molécules vers l'état d'équilibre. Il s'effectue,
normalement, dans l'état vitreux comme une conséquence de
stockage à température ambiante, il se développe
rapidement tant que la température de vieillissement est proche de la
température de transition vitreuse (Tg).
Le vieillissement physique a une influence dramatique sur les
propriétés des polymères, à savoir réduction
de la résistance aux chocs, comme conséquence de l'augmentation
du temps de relaxation de polymère. Des expériences de
vieillissement ont été effectuées sur des
échantillons de PLA de différentes masses moléculaires
(Mv= 5 300, 20 000, 691 000), ces expériences ont
montrées que la diminution de la masse moléculaire de PLA
provoque une augmentation de la magnitude de l'enthalpie de relaxation à
la transition vitreuse (J.-F. Zhang, 2005).
A-I.5.2. Dégradation non-biologique (ou abiotique)
de poly(acide lactique)
Bien que, la biodégradation des matériaux
polymères est définie comme une dégradation de ces
matériaux sous l'effet d'une activité biologique (en particulier
sous l'action des enzymes), elle est souvent amorcée habituellement et
simultanément par une dégradation abiotique (dégradation
mécanique, photodégradation, dégradation thermique et
dégradation chimique).
L'hydrolyse de poly(acide lactique) illustre parfaitement le
mécanisme de dégradation chimique abiotique.
La dégradation de poly(acide lactique) s'effectue en
présence de l'eau, ce dernier provoque l'hydrolyse de la liaison ester
de PLA. La dégradation de PLA, ainsi que d'autres polyesters (PCL, PPC
(polypropylène carbonate)), s'effectue lentement dans un milieu neutre,
mais elle est fortement exprimée dans un milieu basique que dans celui
acide (N. Lucas, 2008).
De Jong et al. (S. J. De Jong, 2001) ont
observé la dépolymérisation de PLA dans un milieu alcalin
(figure 13). Le mécanisme de dégradation peut être
expliquer par une transestérification intramoléculaire. En effet,
le mécanisme s'effectue en deux étapes, la première
étape consiste à une attaque électrophile
(catalysée par la base) de groupement hydroxyle terminal sur le
deuxième groupement carbonyle de la même chaîne ce qui
mène à la formation d'un cycle. La deuxième étape
consiste à l'hydrolyse du cycle de lactide pour donner deux
molécules d'acide lactique, d'une part, et à une
dégradation intramoléculaire de la chaîne de
polymère restante par une attaque alcalin sur le carbone de groupement
ester, suivi par une hydrolyse de la liaison ester, d'autre part. Enfin, une
telle dégradation donne naissance à de nouvelles molécules
de masse moléculaire inférieure à celle de la chaîne
initiale.
Chapitre I. Rappels bibliographiques sur le poly(acide
lactique) et l'ibuprofène

O
O
R
HO
O
HO
O
O
O
O
O
n
O
O
milieu de la chaîne
Poly(acide lactique)n HOextrémité de la
chaîne
Poly(acide lactique)n-x
O
OH
O
HO
O
O
O
O
O
O
R
O
O
O
R
O
O
O
O
n
O
O
O
O
O
O
n
O
HO
H2O H2O
+
O
R
O
O
OH
n
O
Poly(acide lactique)x
O
H2O
O
O
O + O
O
HO
OH
Lactide
O
O
OH
O
O
O
HO
O
O
Poly(acide lactique)n-2
O
OH
Acide lactique Figure 13. Hydrolyse de
poly(acide lactique) dans un milieu alcalin (S. J. De Jong,
2001).
Dans un milieu acide (figure 14), une attaque acide
(H+) sur le groupement hydroxyle terminal de la chaîne de PLA
mène à la formation d'une liaison par pont hydrogène
intramoléculaire. Ensuite, l'hydrolyse de groupement ester permet la
libération d'une molécule d'acide lactique, ce qui fait diminuer
le degré de polymérisation des chaînes de PLA. Enfin, des
réactions de protonation de l'atome de carbone des groupements esters de
la chaîne de PLA conduisent aussi à l'hydrolyse des liaisons
esters correspondantes. Cette hydrolyse donne naissance à
différents fragments de faibles masses moléculaires (S.
J. De Jong, 2001).



H
O
O
O
H


O
O
R
milieu de la chaîne


H
O

R
O
O
OH
O
O
nO


O O
Poly(acide lactique)n
extrémité de la chaîne
OH
O
O
H
O O
O
R
O
O
O
OH
n
O
O
n
O
O
O
O
O H2O
O
O H2O

OH
O
O
O
OH
O
HO
O

R
O
O

O
H

OH
H
O
OH
Acide lactique
OH
O
O
+
OH
O
O
O
O n
O
O
R
O
n
HO
O
O
O
H

R
O
O
O
OH
n
O
Poly(acide lactique)n-x
+
O
H
O
nO
O
O
H
O
O
O
O
O
O
O
Poly(acide lactique)x
Poly(acide lactique)n-1
Figure 14. Hydrolyse de poly(acide lactique)
dans un milieu acide (S. J. De Jong, 2001).
A-I.5.3. Biodégradation de poly(acide lactique)
La biodégradation peut être définie, par
convention, comme « un changement chimique dans le polymère
favorisé par des organismes vivants (souvent des micro-organismes)
». En général, la biodégradation des polymères
se traduit comme un processus extracellulaire (la membrane cellulaire est
imperméable aux macromolécules) catalysé par des enzymes.
Comme la plupart des polymères facilement biodégradables sont
insolubles dans l'eau, la réaction de dégradation doit être
hétérogène et est initialement localisée sur la
surface de polymère. Le processus de biodégradation peut
être schématisé comme suit (schéma 1) (A.
L.
Andrady, 2007).
|
Polymères à haut poids moléculaire
|
|
Polymères à faible poids moléculaire
|
|
Intermédiares organiques
|
|
|
|
|
|
O2
|
|
|
|
|
|

CO2 + H2O + Energie Respiration de la biomasse
Schéma 1. Processus de biodégradation de
polymères biodégradables.
A-I.5.4. Facteurs influençant la
biodégradation de poly(acide lactique)
En général, la dégradation d'un
polymère s'effectue au niveau de sa chaîne principale ou ses
chaînes secondaires. La dégradation des polyesters
biodégradables s'effectue soit par voie chimique ou biologique et dans
certaines étapes de ce processus, les deux voies peuvent être
apparaître au même temps.
Plusieurs facteurs peuvent affecter la biodégradation des
polymères, à savoir :
- Facteurs associés avec la structure de 1er
ordre (structure chimique, masse moléculaire et distribution de la masse
moléculaire) ;
- Facteurs associés avec la structure d'ordre
supérieur (température de transition vitreuse (Tg),
température de fusion (Tf), cristallinité, structure cristalline
et module d'élasticité) ;
- Facteurs reliés aux conditions superficiels
(propriétés hydrophobes et hydrophiles, surface).
En effet, plusieurs études ont montrée que la
partie cristalline de PLA est beaucoup plus résistante à la
biodégradation que la partie amorphe, et que la vitesse de
dégradation diminue avec l'augmentation de la cristallinité. La
masse moléculaire influe sur le processus de dégradation des
polymères. Dans le cas des polyesters, les polymères de masse
moléculaire élevée se dégradent lentement
comparés à ceux de faible masse moléculaire. La
température de fusion des polyesters affecte considérablement
leur dégradation enzymatique. La biodégradation est inversement
proportionnelle à l'augmentation de la température de fusion
(Y. Tokiwa, 2006).
A-I.5.5. Dégradation biologique (ou
biodégradation) de poly(acide lactique)
Il existe dans la nature des microorganismes
(bactéries, levures, champignons, etc.) qui sont responsables de la
dégradation microbiologique des polymères par la
sécrétion d'enzymes ou des produits chimiques (à savoir
acides ou peroxydes). Il y a aussi des microorganismes qui provoquent la
digestion des polymères en causant par la suite un vieillissement
mécanique, chimique ou enzymatique de ces polymères (C.
Bastoli, 2005).
La dégradation de poly(acide lactique) s'effectue dans
un milieu humide et sous des températures élevées. La
dégradation environnementale de PLA s'effectue selon un processus de
deux étapes. D'abord, une hydrolyse lente des chaînes de PLA de
masse moléculaire élevée qui donne naissance à des
oligomères de masse moléculaire plus faible. Ce processus peut
être influencé par certains facteurs à savoir, des acides
ou bases, l'humidité et la

Poly(acide lactique)
température. À la fin de cette première
phase, un changement significatif dans la structure de polymère est
signalé. Ensuite, dans la seconde phase, des microorganismes
interviennent pour poursuivre le processus de dégradation par la
transformation des oligomères de faible masse moléculaire en
dioxyde de carbone (CO2) et l'eau (en présence d'oxygène) ou
méthane (en absence d'oxygène) (J.-F. Zhang,
2005). La figure 15 suivante explique le mécanisme de
biodégradation de PLA :


Biodégradation
M icroorganismes
Enzymes
Oligomères d'acide
lactique Dégradation chimique
Figure 15. Mécanisme de
dégradation de poly(acide lactique) (J.-F. Zhang,
2005).
A-I.5.5.1. Dégradation microbienne de poly(acide
lactique)
Ohkita et Lee, 2006 (T. Ohkhika, 2006), ont
démontré que la dégradation de PLA dans le sol est lente,
en effet, l'enterrement des feuilles de PLA dans le sol pendant 6 semaines n'a
donné aucun signe de dégradation. Le poly(acide lactique) peut
être aussi dégradé dans un compost où il est
hydrolysé en petites molécules (oligomères, dimères
et monomères) à une température variant de 50 à 60
°C pendant 45 à 60 jours. Ces petites molécules sont ensuite
dégradées en CO2 et H2O par des microorganismes demeurant dans le
compost (Y. Tokiwa, 2006).
La dégradation microbienne de poly(acide lactique) a
été réalisée pour la première fois par
Pranamuda et al. (H. Paranamuda, 1997) en utilisant une souche
appelée actinomycete Amycolatopsis isolée à
partir de sol. L'étude de la dégradation de L-PLA au moyen de
cette souche a montrée que pendant 14 jours de culture, 60% de la masse
de polymère était dégradé. Cependant, la souche
Amycolatopsis n'a pas pu métaboliser les produits de
dégradation de PLA. Un autre type de microorganismes,
Amycolatopsis K104-1, a été isolé à partir
de sol. Cette variété de microorganismes pouvait dégrader
plus de 90 % de L-PLA après 8 jours. En plus de la variété
Amycolatopsis, de divers actinomycetes : Lentzea,
Kibdelosporangium, Streptoalloteichus, et
Saccharothrix sont aussi capable de dégrader le LPLA
(Y. Tokiwa, 2006).
La dégradation microbienne de L-PLA à haute
température (= Tg (L-PLA) = 55°C) peut être
obtenue à T = 60 °C en utilisant des microorganismes thermophiles
(Brevibacillus sp). Le Bacillus smithii peut dégrader
35,6 % de la masse de L-PLA à 60 °C pendant 3 jours.
Il a été montré que la présence de
substances naturelles telle que la gélatine dans le milieu de culture
provoque une activité de dégradation élevée de
L-PLA en présence de plusieurs microorganismes. En effet, l'addition de
gélatine dans le milieu de culture de T. album a amélioré
l'activité de dégradation de L-PLA de 76 % par l'intervention de
ce champignon. Les microorganismes, Lentzea waywayandensis et
Kibdelosporangium, sont capables de dégrader plus de 95 % de la
masse des films de PLA de haut poids moléculaire dans un milieu basique
contenant 0,1 % de gélatine pendant 14 jours (Y. Tokiwa,
2006).
A-I.5.5.2. Dégradation enzymatique de poly(acide
lactique)
Les enzymes jouent un rôle important dans le
mécanisme de dégradation des polymères. La
dégradation enzymatique des polyesters aliphatiques par hydrolyse est un
processus qui se fait en deux étapes. La première étape
consiste à l'adsorption de l'enzyme sur la surface de substrat et la
seconde étape consiste en l'hydrolyse des liaisons esters de substrat
(Y. Tokiwa, 2006).
Williams (D. F. Williams, 1981) a
été le premier qui a étudie la dégradation
enzymatique de LPLA au moyen de l'enzyme protéinase de T. album. Par la
suite, Oda Y. et al. (Y. Oda, 2000) ont examiné la
dégradation enzymatique de L-PLA à 50 °C en utilisant 56
protéases commerciales. Cette étude a montrée que les
protéases neutres et acides ont peu ou pas d'effet sur l'activité
de dégradation de L-PLA, mais certaines protéases alcalines
dérivées de Bacillus spp. ont montrés une
activité de dégradation remarquable de L-PLA (Y. Tokiwa,
2006). Enfin, pour montrer la sensibilité de la
dégradation de PLA vis-à-vis des enzymes, une exposition
enzymatique pendant 48 h de PLA pur a peut provoquer une dégradation de
20 % de son poids total (J.-F. Zhang, 2005).
Le tableau 3 suivant résume les différents
microorganismes et leurs enzymes correspondants responsables de la
dégradation de poly(acide lactique).
Tableau 3. Microorganismes responsables de la
dégradation de PLA (Y. Tokiwa, 2006).
|
Microorganisme
|
Type d'enzyme
|
Poly(acide lactique)
|
|
Amicolatopsis sp. (HT 32)
|
Protéase
|
L-PLA
|
|
Amicolatopsis sp. (3118)
|
Protéase
|
L-PLA
|
|
Amicolatopsis sp. (KT-s-9)
|
Protéase
|
L-PLA
|
|
Amicolatopsis sp. (41)
|
Protéase
|
L-PLA
|
|
Amicolatopsis sp. (K104-1)
|
Protéase
|
L-PLA
|
|
Lentzea waywayandensis
|
Protéase
|
L-PLA
|
|
Kibdelsporangium aridum
|
Protéase
|
L-PLA
|
|
Tritirachium album ATCC 22563
|
Protéase
|
L-PLA
|
|
Brevibacillus
|
Protéase
|
L-PLA
|
|
Bacillus stearothermophilus
|
Protéase
|
D-PLA
|
|
Geobacillus thermocatenulatus
|
Protéase
|
L-PLA
|
|
Bacillus sinithii (PL 21)
|
Lipase (esterase)
|
L-PLA
|
|
Paenibacillus amylolyticus (TB-13)
|
Lipase
|
DL-PLA
|
|
Cryptococcus sp. (S-2)
|
Lipase (Cutinase)
|
L-PLA
|
A-I.6. Production industrielle durable de poly(acide
lactique)
Le développement durable est définit comme un
développement qui répond aux besoins des
générations actuelles, sans compromettre la capacité des
générations futures de répondre aux leurs. Le
développement durable est basé sur trois axes principaux qui sont
l'économie, l'environnement et la société (E. T.
H. Vink, 2003).
Récemment, produits, processus et technologie ont
été jugés pour leur durabilité globale. L'analyse
du cycle de vie est l'une des méthodes utilisées pour la
quantification de la durabilité des produits ou processus. Elle prend en
considération plusieurs facteurs : la matière première, le
transport, fabrication du produit, fin d'utilisation et enfin
l'élimination du produit. Autrement dit, l'analyse du cycle de vie
commence à partir de l'extraction de la matière première
utilisée pour la fabrication du produit jusqu'à
l'élimination de ce dernier (i.e. produit) dans les décharges,
incinération ou recyclage (S. Madival, 2009).
Afin de répondre aux besoins du marché
international d'une part et réaliser une production beaucoup plus propre
en respectant les douze principes de la chimie verte d'autre part, une
multitude de société (Tableau 4) ont développé la
production et la qualité de poly(acide lactique) (E.
Aurélie, 2007).
Tableau 4. Les principaux acteurs de la
production de PLA à travers le monde (E. Aurélie,
2007).
|
Logo
|
Nom de polymère
|
Pays
|
Société
|
|
Natureworks®
|
|
Cargill Dow
|
|
|
|
Solanyl®
|
|
Polymix
|
|
|
|
HM® LM®
XM®
|
|
HYCAIL
|
|
|
|
|
Lacea®
|
|
MitsuiChemical
|
|
Galactic®
|
|
|
|
Galactic
|
|
Biophan®
|
|
Treofan
|
|
|
|
Biop®
|
|
Biopolymer Technologies AG
|
|
|
|
La société Cargill Dow basée aux USA,
issue de l'association du groupe agro-alimentaire « Cargill et de
l'industrie chimique Dow chemicals » est leader dans la production des
plastiques à partir des ressources renouvelables. Le projet de
développement de poly(acide lactique) a été lancé
par la société Cargill Inc en 1988. En 1997, le PLA produit par
Cargill Dow a été commercialisé sous le nom commercial
NatureWorksTM (E. T. H. Vink, 2003).
La société Cargill Dow utilise la voie de
polymérisation par ouverture de cycle de lactide pour la production de
poly(acide lactique) (figure 16) (f. E. Henton, 2005).

Figure 16. Processus de préparation sans
solvant de poly(acide lactique) (D. E. Henton, 2005).
Le processus de fabrication de PLA par Cargill Dow est
avantageux pour plusieurs raisons (D.
E. Henton, 2005) :
- Utilisation de 20 à 50 % moins de combustibles fossiles
que les plastiques conventionnels
- Capacité de produire un polymère avec
coût/performance favorable
- Utilisation efficace de la matière première
- Economie d'énergie
- Non utilisation de solvants
- Faible participation à l'émission des gaz
à effet de serre.
Le prix de poly(acide lactique) produit par Cargill Dow LLC
est de l'ordre de 3€/kg fait de lui le polyester biodégradable le
moins cher du marché actuellement. Depuis octobre 2002, Cargill Dow
produit environ 180 000 tonnes de PLA par an (N. E. Suyatma,
2006).
En résumé :
Le poly(acide lactique) est un polyester aliphatique issu
essentiellement des ressources renouvelables. C'est un polymère
biodégradable, biorésorbable et biocompatible connu pour ses
propriétés mécaniques comparables à celles des
polymères pétrochimiques (PE, PS, PP ...). Le poly(acide
lactique) peut être synthétisé par deux voix
différentes : une synthèse directe dite polymérisation par
polycondensation direct de l'acide lactique et à partir d'une
synthèse indirecte dite polymérisation par ouverture de cycle de
lactide.
Le poly(acide lactique) est ses copolymères
possèdent d'excellentes propriétés concernant leurs
applications écologiques, biomédicales et pharmaceutiques.
À cause des raisons suivantes :
i. Production à partir des ressources renouvelables (ex.
maïs, blé, sucre de la betterave) ;
ii. Propriétés mécaniques comparables
à celles des polymères traditionnels, à savoir
résistance mécanique, dureté, rigidité,
élasticité, ainsi que possibilité de traitement par
moulage par injection, extrusion... ;
iii. Dégradabilité de PLA dans le corps humain et
dans l'environnement naturel ;
iv. Très faible toxicité des ses produits de
dégradation (acide lactique et ses oligomères)
dans le corps humain et aussi dans l'environnement naturel
(F. Schwark, 2009).
Aujourd'hui, augmentation de la biodisponibilité,
réduction du coût et la consommation d'énergie, gestion des
déchets et émission de gaz à effet de serre sont en
première priorité dans la stratégie de synthèse de
PLA à l'échelle industrielle. L'année 1997 marque la
naissance d'une nouvelle compagnie, Cargill Dow LLC, destinée à
la production et à la commercialisation de PLA dans le but de
réduire le coût et pour augmenter le volume de la production de
PLA (D. E. Henton, 2005).
Partie B
B-I. Ibuprofène
L'inflammation est un processus biologique de défense
de l'organisme contre un agent agresseur. La thérapeutique
anti-inflammatoire est destinée à contrôler l'excès
de réaction spécifique des tissus et à éviter la
transformation de la phase aiguë de l'inflammation en phase chronique. Les
anti-inflammatoires sont utilisés dans tous les domaines de la
pathologie. Ils appartiennent à des classes chimiques différentes
les unes des autres et sont souvent doués d'une activité
antipyrétique et antalgique périphérique. Le mode d'action
des antiinflammatoires est purement symptomatique.
Les anti-inflammatoires non stéroïdiens (AINS)
regroupent différentes classes chimiques de synthèse de structure
non stéroïdienne, à la différence des
glucocorticoïdes (D. Muster, 2005).
Parmi les AINS les plus utilisés ces dernières
années, on trouve l'ibuprofène. Il a été
découvert pour la première fois par le pharmacologiste Anglais
Stewart Adams au niveau de département de recherche de Boots Pure Drug
Company Ltd à Nottingham (UK). Il a été employé
pour le traitement des douleurs inflammations et de la fièvre. Il a une
large gamme de tolérance et il est souvent plus efficace que l'aspirine
et le paracétamol dans le traitement de plusieurs cas de douleurs. Comme
l'aspirine, paracétamol ou indométhacine, l'ibuprofène est
devenu un standard universel pour la comparaison dans des applications
cliniques et méthodes expérimentales (K. D. Rainsford,
1999).
B-I.1. Identification de l'ibuprofène

OH
O
Figure 17. Structure chimique de
l'ibuprofène (K. D. Rainsford, 1999).
Ibuprofène (IB) est un anti-inflammatoire non
stéroïdien, appartient au groupe des acides de type
2-arylpropanoïque, qui existe sous deux formes énantiomères
R et S. Cependant, seul le composé racémique est utilisé
dans la médecine (K. P. Stock, 1999).
L'ibuprofène est une molécule relativement simple appelée
acide (2RS)-2-[4-(2-méthylpropyl)phényl]propanoïque,
constituée d'un seul groupement fonctionnel (-COOH) et d'une partie
hydrocarbonée inerte (hydrophobe)[ -CH(CH3)C6H4CH2CH(CH3)2] (figure
17).
B-I.2. Chimie médicinale de l'ibuprofène
B-I.2.1. Métabolites de l'ibuprofène
La molécule d'ibuprofène (IB) ne possède
pas de groupements facilement hydrolysables comme amide et ester. Des
études ont montré que l'IB présente deux principaux
métabolites, désignées par A et B, dans l'urine d'un homme
normal (figure 18). Les deux métabolites ont été
facilement isolés. Il a été remarqué que la
molécule d'IB a conservé son squelette carboné initial
ainsi que le groupement carboxylique.
COOH


COOH
COOH
OH
Figure 18. Structure chimique des deux
principaux métabolites de l'ibuprofène (K. D. Rainsford,
1999).
Les deux métabolites A et B qui sont originellement
trouvées dans l'urine de l'homme ont été plus tard
détectées dans l'urine de rat, babouin et chien en
quantités variables. En 1969, deux autres métabolites avaient
été détectées (figure 19).

COOH
OH
Figure 19. Structure chimique des deux autres
métabolites de l'IB (K. D. Rainsford, 1999).
B-I.2.2. Enantiomères de l'ibuprofène
La molécule d'ibuprofène possède un
centre chiral au niveau de groupement acide propanoïque. Par
conséquent, deux isomères optiques sont possibles : la forme (+)
dextrogyre ou (S)-(+)-ibuprofène et la forme (-) lévogyre ou
(R)-(-)-ibuprofène (figure 20).
R-(-)-ibuprofène S-(+)-ibuprofène
Figure 20. Structure chimique des deux formes
énantiomèriques de l'IB (K. D. Rainsford,
1999).
Les deux isomères optiques de l'ibuprofène
présentent des effets biologiques différents. Par
conséquent, l'isomère S-(+) a connu un grand intérêt
dans le domaine de la médecine car il est pharmacologiquement actif
comme inhibiteur de la synthèse des prostaglandines, et l'isomère
R-(-) qui est moins actif que S-(+) dans l'inhibition de la synthèse des
prostaglandines, mais il peut avoir certains propriétés
pharmacologiques reliées à l'action anti-inflammatoire de l'IB
(K. P. Stock, 1999). Comme chacun des deux
énantiomères présente une activité pharmacologique
importante, il est nécessaire donc d'avoir les deux formes
séparément. Pour se faire, il existe deux types d'approches : la
première consiste à la séparation de mélange
racémique d'IB et la deuxième approche consiste à la
synthèse de chaque énantiomère tout seul par une
méthode de synthèse dite stéréosélective
(K. D. Rainsford, 2009).
B-I.3. Pharmacie de l'ibuprofène
B-I.3.1. Caractéristiques de l'ibuprofène
B-I.3.1.1. Caractéristiques physiques et chimiques
de l'ibuprofène
L'ibuprofène est disponible sous forme d'une poudre
blanche cristallisée. C'est un solide faiblement cireux avec une faible
odeur et un goût fort et caractéristique. Une fois avalé,
l'IB laisse une sensation brûlante dans la gorge. Comme les
méthodes de synthèse de l'ibuprofène sont nombreuses, les
impuretés suivantes (tableau 5) peuvent apparaitre dans le produit
final.
Tableau 5. Différentes impuretés
détectées dans l'ibuprofène (K. D. Rainsford,
1999).
|
Impureté
|
Quantité (ug/g)
|
|
Acide 2-(4-méthylphényl)propanoïque *
|
Jusqu'à 100
|
|
2-(4-isobutylphényl)propioamide *
|
1000
|
|
Acide 2-(4-n-propylphényl)propanoïque *
|
Jusqu'à 500
|
|
Acide 2-(3-isobutylphényl)propanoïque *
|
500
|
|
Acide 2-(4-n-butylphényl)propanoïque *
|
Jusqu'à 500
|
|
Acide 2-hydroxy-2-(4-isobutyl phényl) propanoïque
**
|
< 500
|
|
Acide 2-(3-isobutylphényl)propanoïque **
|
~ 600
|
|
Acide 2-(4-n-butylphényl)propanoïque **
|
3000
|
|
Acide di-isobutylisotropique **
|
< 500
|
|
1,3-di-(isobutylphényl)butane **
|
< 200
|
|
1,3-di-(isobutylphényl)-1-butanone **
|
< 200
|
NB. * fournisseur : Knoll Pharmaceuticals, ** fournisseur :
Albermale Corporation.
L'ibuprofène est faiblement soluble dans l'hexane, mais
soluble dans l'éthanol, octanol, diméthylsulfoxyde et
chloroforme. Le tableau 6 suivant donne la solubilité approximative de
l'IB dans certains solvants organiques.
Tableau 6. Solubilité de
l'ibuprofène dans des solvants organiques (K. D. Rainsford,
1999).
|
Solvant
|
Solubilité approximative à
température ambiante (%)
|
|
Acétone
|
> 10
|
|
Ethanol
|
> 10
|
|
Octanol
|
33.0
|
|
Hexane
|
3,3
|
|
Eau distillée
|
< 0,1
|
La solubilité de l'ibuprofène est
proportionnelle au pH du milieu (figure 21). En effet, la solubilité de
l'IB augmente brusquement avec le pH, c-à-d le médicament
étant en grande partie insoluble à de faibles pH, il devient
facilement soluble à des pH alcalins.

Figure 21. Solubilité de
l'ibuprofène en fonction du pH (K. D. Rainsford,
1999).
L'ibuprofène est une poudre essentiellement
non-hygroscopique, car des expériences de stockage de celle-ci dans des
endroits de différents pourcentages d'humidité (0, 31, 58, 86, 94
et 100 %) pendant 3 mois ont montré que la masse de l'IB reste
constante.
Les caractéristiques physiques (comme la densité)
de l'ibuprofène sont liées au paramètre taille des
particules comme le montre tableau 7 suivant.
Tableau 7. Caractéristiques physiques
(K. D. Rainsford, 1999).
|
Catégories disponibles
|
IB25
|
IB38
|
IB50
|
|
Taille des particules (um)
|
20-33
|
33-45
|
45-60
|
|
Densité volumique (g/cm3)
|
0,20-0,40
|
0,25-0,50
|
0,40-0,60
|
|
Densité tapée (g/cm3)
|
0,40-0,60
|
0,50-0,70
|
0,60-0,80
|
Source Knoll Pharmaceuticals
Les différentes caractéristiques chimiques de
l'ibuprofène sont résumées dans le tableau 8 suivant
(d'après la Pharmacopée européenne).
Tableau 8. Caractéristiques chimiques de
l'ibuprofène (K. D. Rainsford, 1999).
|
Test
|
Caractéristique
|
|
Aspect en solution
|
Clair, non colorée
|
|
Rotation optique
|
-0,05 à 0,05 °
|
|
Métaux lourds
|
Au maximum 10 ug/g
|
|
Perte à la dessiccation
|
Au maximum 5000 ug/g
|
|
Cendre sulfurique
|
Au maximum 1000 ug/g
|
|
Eau
|
Au maximum 1%
|
|
Quantité de 4-isobutyrylacétophénone *
|
Au maximum 1000 ug/g
|
* c'est le produit de dégradation majoritaire de
l'ibuprofène.
B-I.3.1.2. Caractéristiques physicochimiques de
l'ibuprofène
Des études utilisant la méthode
calorimétrique différentielle (DSC) ont montré que le
degré de cristallinité et le solvant utilisé pour la
cristallisation de l'ibuprofène ont un effet sur son point de fusion
(S. Lerdkanchanaporn, 1997). Romero et al. (A. J.
Romero a, 1993 ; A. J. Romero b, 1993), ont
suggéré que l'aspect stéréochimique de
l'ibuprofène affecte ses propriétés et sa forme
cristalline. Ils ont montré que l'isomère (+)-IB possède
un point de fusion plus faible que celle l'isomère (-)-IB. Le point de
fusion de (+)-IB et (-)-IB est aux environ de 47-54 °C alors que le
composé racémique possède un point de fusion aux environ
de 76-78 °C. Quelques autres caractéristiques physicochimiques de
l'ibuprofène sont résumées dans le tableau 9 suivant.
Tableau 9. Quelques propriétés
physicochimiques de l'IB.
|
Masse molaire
(g/mol) (a)
|
pKa
(a)
|
ëmax (nm)
(a)
|
ÄHfus
(KJ/mol)
(a)
|
ÄHsub
(KJ/mol)
(b)
|
ÄHvap
(KJ/mol)
(b)
|
|
206,27
|
5,2
|
265
|
25,5
|
121
|
42,7
|
a) (Y. J. Manrique, 2008) (b) (A. J.
Romero, 1993)
B-I.3.1.3. Caractéristiques cristallographiques de
l'ibuprofène
L'ibuprofène se cristallise avec deux molécules
par unité asymétrique pour former un dimère cyclique au
moyen de liaisons par pont hydrogène. Les deux molécules formant
le dimère peuvent être de la même nature (i.e. S-(+)/S-(+))
ou de natures différentes (i.e. mélange racémique :
R-(-)/S-(+)), la figure 22 montre les deux cas.

R =
Dimère R/S (racémique)
O H O
R
O H O
R
R
R
O
H
O
O H O
Dimère S/S
Figure 22. Formation d'un dimère
cyclique entre deux molécules d'IB (A. A. Freer,
1993).
Des caractéristiques cristallographiques de la maille des
cristaux d'ibuprofène sont données dans le tableau 10 suivant.
Tableau 10. Donnée cristallographiques
sur la maille d'ibuprofène (A. A. Freer, 1993).
|
Type de réseau
|
Groupe spatial
|
Paramètres de maille
|
Volume de la maille
|
Nombre de motif /maille(Z)
|
|
Monoclinique
|
P21/c
|
a = 12,462 A
b = 8,035 A
c = 13,539 A â = 112,89 °
|
1248,8 A3
|
4
|
B-I.3.2. Disponibilité de l'ibuprofène dans
le monde
Malgré la sophistication relative du marché de
l'ibuprofène, les formes galéniques sous forme de
comprimés sont toujours prédominantes. Cependant, des capsules,
sirops oraux et des préparations topiques sont aussi disponibles. Le
tableau 11 suivant montre la distribution des formes galéniques de
l'ibuprofène dans le marché européen et au Etats-Unis.
Tableau 11. Disponibilité des formes
galéniques dans l'Europe et les Etats-Unis (%) (K. D. Rainsford,
1999).
|
Forme galénique
|
Royaume- uni
|
France
|
Allemagne
|
Italy
|
Pays- bas
|
Belgique
|
Espagne
|
USA
|
|
Comprimé
|
82
|
95
|
76
|
88
|
98
|
89
|
89
|
97
|
|
Effervescent
|
0,3
|
/
|
0,3
|
/
|
/
|
0,6
|
/
|
/
|
|
Capsule
|
/
|
4
|
1
|
/
|
/
|
0,6
|
6,5
|
1
|
|
Toute forme à libération
contrôlée
|
0,5
|
/
|
/
|
2,5
|
/
|
/
|
/
|
/
|
|
Liquide oral
|
2
|
/
|
/
|
/
|
1,3
|
/
|
/
|
2,1
|
|
Topique
|
16
|
/
|
21
|
1,5
|
0,4
|
10
|
/
|
/
|
B-I.4. Pharmacocinétique de l'ibuprofène
B-I.4.1. Mécanisme d'action
Comme tous les anti-inflammatoires conventionnels,
l'ibuprofène diminue la synthèse des prostaglandines par
l'inhibition sélective de la cyclo-oxygénase (COX).
L'activité inhibitrice de l'IB est due à
l'énantiomère S-(+) (I. Tegeder, 2001).
Toutefois, l'énantiomère R-(-) inactif peut
être bio-converti en la forme active S-(+). En effet, environ 40 à
60 % de la forme R-(-) de l'IB est métaboliquement converti à la
forme S(+) au niveau de la partie gastro-intestinal et le foie après
absorption orale. La première étape implique l'activation de
R-(-)-ibuprofène avec ATP Mg pour former la dérivée AMP
qui est ensuite estérifié avec le coenzyme A par l'action de
acyl-CoA synthétase. Le R-ibuprofèneCoA subit une
épimérisation pour donner S-ibuprofène-CoA qui est ensuite
hydrolysé pour former S-ibuprofène, comme il est montré
dans la figure 23.
La cyclo-oxygénase existe sous deux iso-formes COX-1 et
COX-2. Les eicosanoïdes produits par la COX-1 protègent la muqueuse
gastrique, régulent le débit de la filtration glomérulaire
et induisent l'agrégation plaquettaire, et la COX-2 agit dans
l'inflammation et la douleur par la production de prostaglandines
vasodilatatrices. L'inhibition de la COX-2 par l'IB explique son effet
antipyrétique et analgésique (E. Autret-Leca,
2006).
En plus, l'ibuprofène possède des
propriétés indépendantes de l'inhibition des
prostaglandines. Parmi eux, l'inhibition de facteurs de transcription (comme
NF-kappa-B et AP1), qui contribueraient à l'activité
anti-inflammatoire. Ces facteurs de transcription interviennent notamment dans
la production de diverses cytokines pro-inflammatoires et dans la maturation
des cellules immunitaires (I. Tegeder, 2001).
H H

COSCoA
COO
R-ibuprofène R-ibuprofène CoA

Intermédiaire planaire

CH3
COSCoA
S-ibuprofène CoA
COO
S-ibuprofène
H
Figure 23. Conversion de
R(-)-ibuprofène en S(+)-ibuprofène au moyen d'une activité
catalytique de l'acyl
coenzyme thioestérase (K. D. Rainsford,
2009).
B-I.4.1. Absorption de la molécule
d'ibuprofène dans l'organisme
L'ibuprofène est bien absorbé par le tube
digestif. Il est rapidement absorbé dans la partie gastro-intestinal
supérieure, et le pic plasmatique des énantiomères R et S
est observé 1 à 2 heures environ après administration par
voie orale (K. D. Rainsford, 2009).
La plupart des formulations orales de l'ibuprofène
montrent une biodisponibilité systémique complète,
independemment de leur absorption. La biodisponibilité absolu des
énantiomères R-(-) et S-(+) de l'IB est égale à
83,6 % et 92,0 %, respectivement. En général, le taux
d'absorption, tmax et Cmax dépend de la
formulation (H. Cheng, 1994).
La figure 24 montre les profiles d'absorption de
l'ibuprofène dans l'organisme humain.

R-(-)-IB après IB racémique R-(-)-IB apès
R-(-)-IB S-(+)-IB après S-(+)-IB
Temps (heure)
Figure 24. Profiles d'absorptions cumulatifs
moyens des énantiomères de l'IB après une administration
séparée
de l'IB racémique et de chaque
énantiomère (H. Chang, 1994).
B-I.4.2. Distribution de la molécule
d'ibuprofène dans l'organisme
L'ibuprofène est lié à 99 % à
l'albumine plasmatique et possède un faible volume de distribution. Dans
le liquide synovial, il peut atteindre des concentrations supérieures
aux concentrations sériques (N. M. Davier, 1998). Dans
le fluide synovial, tmax des deux énantiomères de
l'ibuprofène est approximativement 2 heures. La distribution de
l'ibuprofène dans les tissus humains est disponible à partir des
études in vitro. Ces études ont montré que
l'ibuprofène peut se lier aux tissus de la peau, des muscles,
sous-cutanés et tendons. Cependant, cette liaison est plus importante
avec les muscles, mais elle est moins importante avec les tissus tendons. Il a
été montré aussi que des concentrations
élevées de l'ibuprofène étaient
détectées dans le foie et dans le sang (K. D. Rainsford,
1999).
B-I.4.3. Demi-vie et élimination de
l'ibuprofène dans l'organisme
L'ibuprofène possède une demi-vie courte,
située entre 2 et 4 heures chez l'homme et le chien. Cette
période de demi-vie est prolongée en cas d'insuffisance
rénale et hépatique.
L'excrétion de l'ibuprofène est souvent
complète dans 24 heures. Il est excrété, de 50 à 60
% sous sa forme métabolisée et d'environ 10 % sous sa forme
d'origine (ou inchangée), principalement au moyen de filtration
glomérulaire et sécrétion tubulaire. Le reste de
médicament est éliminé sous forme d'un résidu
solide (métabolites et médicaments non absorbés)
(J. A. Richardson, 2001).
L'excrétion de l'ibuprofène dans le lait des
seins est négligeable ; seulement environ 1 mg d'une dose
racémique de 400 mg est excrété par jour dans le lait.
Ceci peut être attribué à
l'acidité élevée de l'ibuprofène et
aussi à sa liaison avec les protéines plasmatiques. La
sécrétion de l'IB dans la salive peut être aussi
négligeable (K. D. Rainsford, 1999).
B-I.5. Utilisation thérapeutique de
l'ibuprofène
L'ibuprofène est connu pour ses
propriétés anti-inflammatoires, antipyrétiques et
analgésiques qui assurent un effet thérapeutique rapide. Il est
habituellement commercialisé sous forme d'ibuprofène
racémique. Il est disponible sous forme de comprimés de 50, 100,
200, 300, 400, 600 et 800 mg (J. A. Richardson, 2001).
L'ibuprofène est disponible sur ordonnance (ex.
Brufen®), en général à des doses allant
jusqu'à 3200 mg par jour, principalement pour le traitement des
affections douloureuses et anti-inflammatoires, notamment la polyarthrite
rhumatoïde, la spondylarthrite ankylosante, l'arthrose, les douleurs
postopératoires, les douleurs post-pentum et lésions des tissus
mous. Il est également disponible en tant que médicament sans
ordonnance (ex. Nurofen®), en général à des doses
allant jusqu'à 1200 mg par jour, principalement pour le traitement des
symptômes de douleur et de fièvre, notamment
céphalée, migraine, névralgie, dysménorrhée,
douleur dentaire et rhume et grippe. L'ibuprofène et ses
dérivés sont également proposés pour d'autres
usages thérapeutiques, notamment pour le traitement de la perte osseuse
parodontale du prurit et de la maladie d'Alzheimer (G. Plucker,
2004). À cause de ses propriétés
antiprolifératives et proapoptotiques, l'ibuprofène pourrait
avoir un usage clinique pour la chimio-prévention de certains cancers
(R. K. Yeh, 2004). Il a été aussi
suggéré que l'ibuprofène puisse réduire la
mortalité en cas de septicémie associée à une
hypothermie (M. M. Arons, 1999).
B-I.6. Effets indésirables et intoxication de
l'ibuprofène
L'ibuprofène a été introduit aux
Etats-Unis en 1974 comme un médicament délivré sur
ordonnance, mais dés 1984 il est devenu disponible sans ordonnance. Par
conséquent, la dose de l'ibuprofène a augmentée dés
sa délivrance sans ordonnance (ou en vente libre)
(W.
Holubeck, 2007).
La concentration thérapeutique normale de
l'ibuprofène dans le sang est dans l'ordre de 50 mg/dm3, mais
à des concentrations voisines de 250 mg/dm3 il devient
toxique (M. Melillo, 2004). Le profile toxicologique de
l'ibuprofène est bien reconnu par l'apparition de divers effets
cliniques, à savoir hémorragie gastro-intestinale,
dépression de système central nerveux, problèmes
respiratoires, insuffisance rénale aiguë, toxicité
hépatique, thrombopénie, hypothermie et la mort (W.
Holubeck, 2007 ; M. Melillo, 2004).
L'ibuprofène ainsi que d'autres AINS peuvent provoquer
d'autres effets secondaires plus ou moins importants, citant nausée,
vomissement, diarrhée, brûlure de l'estomac, douleur de
l'épigastre, douleur abdominale, dyspepsie, flatulence et constipation,
hypertension, contraction des myocardes, agranulositose, anémie,
méningite aseptique et réaction anphilactique et réaction
cutané (K. D. Rainsford, 2009).
B-I.7. Utilisation de l'ibuprofène dans les
systèmes à libération prolongée
Grace à ses propriétés pharmacologiques,
à savoir le temps de la concentration maximale (Tmax= 1,5 h) et la
période de demi-vie (t1/2 = 2 h), l'ibuprofène est
considéré comme étant un
médicament idéal (ou modèle) pour le
développement des systèmes à libération
prolongée de médicaments.
Comme c'est déjà mentionné dans ce
chapitre, l'ibuprofène sous forme de comprimés est la forme
posologique prédominante sur le marché. L'ibuprofène
présente des caractéristiques de compressibilité
modérées comparativement à celles des autres AINS (ex.
paracétamol). Par conséquent, et dans le but de prolonger la
libération de l'IB, des comprimés à base
d'ibuprofène ont été préparés au moyen de
plusieurs techniques. Ces méthodes impliquent souvent l'enrobage direct
de principe actif par des excipients, et l'incorporation du principe actif
à l'intérieur d'une matrice (ex. polymère) qui peut
libérer le principe actif sous l'effet d'un processus d'érosion
et/ou diffusion (K. D. Rainsford, 1999).
Ho et Blank (V. T. R. Ho, 1989), ont
préparé des comprimés à base d'ibuprofène
contenant de l'amidon pré-gélifié ainsi que d'autres
agents mouillant par la technique de séchage par atomisation (ou
pulvérisation). Ces comprimés ont montré une
stabilité, biodisponibilité et dissolution
améliorées. La température de fusion relativement faible
de l'ibuprofène (75 °C) était exploitée par certains
chercheurs pour produire des formulations pharmaceutiques solide. Deboeck et
col. (A. M. Deboeck, 1990), ont exploité cette
dernière (i.e. Tf faible) pour préparer un eutectique à
partir d'ibuprofène et des acides gras, cet eutectique a
été ensuite introduit dans des gélules (capsules en
gélatine). Deux groupes de chercheurs de la compagnie Anglaise Boots ont
utilisé des techniques de cristallisation pour produire des
agglomérats d'ibuprofène avec l'amidon qui possédaient de
bonnes propriétés de compression Atkin et al.
(G. J. Atkin, 1993).
A. Fernández-Carballido et al. (A.
Fernández-Carballido, 2004), ont préparé des
microsphères à base de copolymère PLGA Chargées
d'ibuprofène par la méthode de microencapsulation à partir
d'une émulsion huile/eau. Ces formulations sont capables de
libérer l'ibuprofène à 24 ug/h sur une période de
temps la plus longue possible et sont destinées à une
administration intra-articulaire. Des nanoparticules à base de
polymère DEAE dextran (Ddex) chargées d'IB ont été
préparées par le groupe de chercheurs Bingbing Jiang et al.
(B. Jiang, 2005) en utilisant la méthode de
co-précipitation. Les nanoparticules préparées ont
montrées certains avantages, à savoir remplissage positive des
nanoparticules en IB, stabilité lors de stockage grâce au
phénomène de coagulation entre les nanoparticules et la
libération prolongée de l'IB à partir des nanoparticules.
Ont trouve aussi le travail du groupe C. J. Thompson et al. (C. J.
Thompson, 2007) sur la préparation des microsphères
à base de nouveaux polyesters chargées d'ibuprofène. Ces
microsphères ont été préparées par la
méthode d'évaporation de solvant. L'ibuprofène
était choisi comme médicament hydrophobe modèle à
cause de sa tendance à former des conjugués en présence
des polyesters utilisés. Les microsphères ainsi
préparées ont montrées une efficacité
d'encapsulation élevée et une libération prolongée
d'IB qui dépend de plusieurs facteurs, efficacité
d'encapsulation, solubilité de principe actif, surface et morphologie
des microsphères, nature hydrophobe et cristallinité du
polymère.
En résumé :
L'ibuprofène est un médicament prototype de la
famille des anti-inflammatoires non stéroïdiens, très
utilisé dans le monde entier à cause de ses
propriétés antiarthritiques, analgésiques et
antipyrétiques (K. D. Rainsford, 1999). Les
propriétés pharmacologiques de l'ibuprofène sont
nombreuses, à savoir :
i. Volume de distribution faible de l'IB dans l'organisme comme
conséquence de sa liaison avec les protéines plasmatiques
(albumine) ;
ii. Capacité de s'accumuler en quantité
appréciable dans les compartiments enflammés où une
activité anti-inflammatoire et analgésique est nécessaire
;
iii. L'ibuprofène possède un temps de demi-vie
relativement faible (2 h) et le
temps de concentration maximale est entre 1 et 2 heures
(K. P. Stock, 1999).
Le mode d'action de l'ibuprofène dans l'organisme
implique l'inhibition de la cyclo- oxygénase (OX-1 et OX-2) par
l'intermédiaire de l'énantiomère S-(+)IB et CoA.
L'ibuprofène possède un potentiel considérable dans le
traitement de nombreuses maladies inflammatoires chroniques et
dégénératives ainsi que dans le traitement de certains
cancers.
L'utilisation abusive de l'ibuprofène dans le
traitement des douleurs pourrait être la cause de la diminution de sont
efficacité apparente et par conséquent, des signes de
toxicité gastro- intestinales ou rénales peuvent être
détectés (K. D. Rainsford, 1999).
Chapitre II. Vectorisation de principes
actifs
Chapitre II : Vectorisation de principes actifs
II.1. Historique
Depuis des décennies, des composés
pharmacologiques n'ont pas cessé d'être découverts,
isolés et synthétisés. Cependant, à cause de la
difficulté de trouver un mode d'administration approprié, un
certains nombre de ces composés prometteurs sont restés à
l'échelle de laboratoire. Parmi ces composés, on trouve, par
exemple, des composés hautement lipophiles, comme le
tétrahydrocannabinol, principe actif du cannabis, et d'autres agents
pharmacologiques comportant des structures moléculaires géantes
comme les peptides, les protéines, les gènes et les
nucléotides (O. Hung, 2006).
Depuis les années 40, des polymères
synthétiques ont été utilisés comme
thérapeutiques. En effet, au niveau clinique, ces polymères
synthétiques étaient utilisés comme plasma expanders (ex.
PVP, Dextran), comme matériels de pansement et comme antiseptiques
(PVPiode appelée povidone). À partir de 1949, le nombre de
publications traitant sur les polymères thérapeutiques «
Polymer therapeutic » a beaucoup augmenté et atteint plus de 6000
articles publiés en 2008 (M. J. Vincent, 2009).
Aux alentour des années 50, c'est la découverte
de la microencapsulation. La microencapsulation peut être physique ou
chimique. Aujourd'hui, le mécanisme d'encapsulation est présent
dans 65% des systèmes à libération contrôlée
(T. Zecheru, 2008).
À partir des années 60, un grand nombre de
chercheurs ont travaillé pour concevoir les premiers conjugués
polymère synthétique-principe actif. La découverte des
systèmes de vectorisation de principes actifs au moyen de
polymères a été publiée en 1964 par Folkman et Long
(J. Folkman, 1964). Cette étude a montrée que
des principes actifs de faibles masses moléculaires peuvent
diffusées à travers les parois d'un tube en silicone à une
vitesse contrôlée. À partir de là, les
polymères ont occupé une place très importante dans le
domaine de la libération contrôlée de principes actifs.
Durant ces dernières années, des systèmes de vectorisation
de médicaments ont été conçus pour réaliser
une libération contrôlée. Ces systèmes
étaient principalement sous forme de microsphères, films,
comprimés, ou dispositif d'implantation (M. J. Vincent, 2009 ;
A. V. Kabanov, 2008).
Depuis les années 70, Langer et Folkman (R.
Langer, 1976) ont montré l'utilité thérapeutique
de la libération contrôlée des médicaments, ce
concept a eu un impact important dans toutes les branches de la
médecine, ainsi que dans d'autres domaines comme l'agriculture
(libération contrôlée de pesticides et de fertilisants),
l'alimentation et les fragrances (J. Jean-François,
2004). Durant cette période aussi, des systèmes de
vectorisation des médicaments implantables ont été
développés pour remplacer les systèmes traditionnels,
comme les pilules et injection hypodermique. Parmi les implants disponibles
actuellement, on trouve les norplant et divers pompes, comme la pompe
d'insuline. Il existe aussi des pompes implantables qui ont été
développées pour le traitement de divers maux, à savoir le
diabète et le cancer. Ces pompes servent à réduire le
besoin des doses de l'insuline répétées ou des injections
de la chimiothérapie. Elles peuvent aussi fournir des prises de sang
pour analyse sans utiliser les systèmes de prélèvement
traditionnels (T. Zecheru, 2008).
Durant les années 80, les systèmes de
vectorisation des médicaments transdermiques commence à
apparaitre. Le premier système transdermique disponible dans le commerce
est le transderm scop qui libère le principe actif scopolamine. Il a
été suivi par les timbres autocollants contenant la
nitroglycérine qui aide à la prévention de l'angine
(T. Zecheru, 2008).
Depuis la fin des années 80 jusqu'à
présent, l'activité immense et le succès clinique des
nano-thérapeutiques est dû à trois facteurs principaux. Le
premier était le concept de « PEGylation » (conjugué de
polyéthylène glycol-principe actif), le second facteur
était le concept « ciblage actif » du principe actif, et enfin
le troisième facteur était le concept « ciblage passif
» qui a été découvert par Hiroshi Maeda (A.
S. Hoffman, 2008) (Japan), où des vecteurs de
nano-échelles ont été enfermés dans les tumeurs.
Récemment, comme un résultat de
développement de la nanotechnologie, une nouvelle
génération de polymères a émergé, elle
utilise des matériaux et des dispositifs à l'échelle de
nanomètre pour transporter des peptides, des médicaments, des
gènes et des oligonucléotides. Ces matériaux vecteurs
englobent des liposomes, dendrimères, micelles polymériques, les
complexes polymère-ADN et d'autres nanoparticules. Cette nouvelle
génération de vecteurs peuvent protéger les agents actifs
de la dégradation dans les liquides biologiques et favorise la
pénétration intracellulaire des substances actives. Ces vecteurs
assurent aussi le transport des agents actifs vers leurs sites d'action
(O. Hung, 2006 ; A. V. Kabanov, 2008).
Plus récemment, c'est le développement des
vecteurs de médicaments qui utilisent des nanoparticules composés
de polymères biodégradables. Ces systèmes peuvent se
présenter sous forme d'une matrice polymère (dite
nanosphère) ou sous forme d'un système réservoir dans
lequel un milieu huileux ou aqueux est entouré d'une membrane
polymérique (nanocapsule). Ce type de vecteur présente certains
avantages comparativement aux liposomes à cause de leur stabilité
pendant le stockage et dans le corps vivant (i.e. in vivo)
(O.
Hung, 2006).
II.2. Concept général de la vectorisation de
principes actifs
Lorsque un principe actif est administré, seul une
très petite fraction de la dose arrive réellement à
atteindre les récepteurs ou les sites d'action appropriés, alors
que le reste de la dose se trouve gaspillé un peut partout entre le
point d'administration et le site d'action dans l'organisme vivant (T.
Allen, 2004).
Pour minimiser la dégradation et la perte de masse du
principe actif, prévenir les effets secondaires indésirables ou
nocifs, augmenter la biodisponibilité et l'accumulation du principe
actif dans la zone pathologique divers systèmes de vectorisation de
principes actifs ont été développés (V. P.
Torchilin, 2004).
La réussite des systèmes de vectorisation de
médicaments nécessite une collaboration entre plusieurs
spécialistes de domaines différents qui sont les chimistes, les
physiologistes et les ingénieurs biomédicaux (T. Zecheru,
2008 ; L. Y. Qiu, 2006).
Le concept de la libération contrôlée
regroupe tout système exerçant un contrôle soit spatial,
soit temporel ou bien les deux à la fois sur la libération d'un
principe actif. Le contrôle temporel signifie le choix d'une
cinétique de libération prédéterminée durant
le traitement. Le contrôle spatial a comme objectif de diriger un
principe actif précisément vers son site d'action
désiré (A. V. Kabanov, 2008 ; L. Y. Qiu,
2006).
La vectorisation de principes actifs est devenue une science
très demandée car les systèmes à libérations
contrôlées comportent plusieurs avantages comparativement aux
systèmes classiques, à savoir :
v' Prolongement de la durée d'action par le
piégeage du principe actif dans la matrice vecteur (C. M. Perry,
1996) ;
v' Diminution de la dose requise pour l'action, et
amélioration de confort du patient (J. Jean-François,
2004) ;
v' Vectorisation localisée du principe actif, en
d'autre terme le vecteur peut être un système à
libération ciblée. Dans ce cas le principe actif peut être
dirigé directement vers le site où il doit intervenir. Pour cela,
les vecteurs en questions sont conçus afin de ne libérer les
principes actifs que sous l'effet de certains facteurs précis (pH,
Température,...). De tels systèmes sont destinés
spécialement à la vectorisation des principes actifs toxiques qui
sont à l'origine de divers effets secondaires (comme les principes
actifs chimiothérapeutiques : ex. les gènes) (T. Zecheru,
2008, H. Laroui, 2007, R. Kircheis, 2002) ;
v' Stabilisation du principe actif, le polymère peut
protéger le principe actif dans le milieu physiologique et par
conséquent l'amélioration de sa stabilité in vivo. Ce type
de vecteurs est particulièrement intéressant pour la
vectorisation des principes actifs de faibles durées de vie (T.
Zecheru, 2008, H. Laroui, 2007) ;
v' Systèmes à libération
contrôlée sont conçus pour véhiculer des principes
actifs à une vitesse spécifique, durant une période de
temps bien définie, ce qui a pour but le maintient de son niveau de
concentration dans la plage thérapeutique comme le montre la figure
25.

Cocentration au niveau du site d'action
Niveau toxique
Plage thérapeutique
Injection
Système à libération
contrôlée
Niveau d'efficacité minimum
heure
Injections chaque 6 heures
Système à libération contrôlée
admistré à 0 heure
Figure 25. Plage thérapeutique d'un
principe actif : Application par un système à libération
contrôlée de
principes actifs comparé à un
système d'administration par injection (J. Khandare,
2010).
II.3. Différentes voies d'administration des
médicaments
Les systèmes à libération
contrôlée sont bénéfiques vis-à-vis de la
santé humaine grâce aux avantages cités
précédemment. Mais plusieurs médicaments présentent
un comportement indésirable dont l'origine est reliée
précisément à la voie d'administration du
médicament (G. W. Cleary, 1984). Le choix de la voie
d'administration du médicament dépend de plusieurs facteurs,
à savoir l'acceptabilité du patient, les propriétés
du principe actif (sa solubilité), l'accès à l'endroit de
la maladie (T. Zecheru, 2008). La figure 26 présente
les différentes voies d'administrations de médicaments.

Figure 26. Différentes voies d'administrations
(M. Mort, 2000).
II.3.1. Voie transdermale
La voie d'administration transdermale a été
utilisée aux USA depuis les années 50 (G. W. Cleary,
1984). La perméation transdermale (ou absorption
percutané) désigne le passage du principe actif de
l'extérieur de la peau vers les diverses couches de cette
dernière et enfin vers le système circulatoire. Le
mécanisme d'administration transdermale de médicaments peut
être schématisé comme le montre la figure 27.

Site de dépôt
4
Capillaires sanguin
Site
d'action
Système de vectorisation transdermale
Stratum corneum Epiderme
Derme
Site de métabolites
Elimination
Figure 27. Processus de perméation
transdermale (T. Higuchi, 1978).
La voie d'administration transdermale présente des
avantages et des inconvénients (G. W. Cleary, 1984 ; A. F.
Kidonieus, 1987). La vectorisation par voie transdermale
possède plusieurs avantages citant parmi eux, elle :
- Evite l'agressivité chimique de la partie
gastro-intestinal (ex. le fluide gastrique), - Assure une absorption
adéquate de certains principes actifs,
- Donne un confort aux patients,
- Assure une utilisation efficace des principes actifs de courte
demi-vie,
- Peut interrompre le passage des principes actifs toxiques
dès l'entrée.
Cette voie d'administration présente aussi certains
inconvénients qui sont :
- le système se limite à l'administration des
principes actifs de faible dose,
- les Adhésifs utilisés ne peuvent pas
s'adhérés à n'importe quelle type de peau,
- la Formulation des médicaments peuvent être
irritantes ou allergiques vis-à-vis de la peau, - les Dispositifs
utilisés peuvent être inconfortables durant l'usage,
- Certains patients ne peuvent pas utiliser ces systèmes
car ils sont chers.
II.3.2. Voie oral
L'administration du principe actif par la voie oral a
été, généralement, la voie la plus commode et la
plus utilisée (J. Jean-Francois, 2004 ; G. S. Kwon,
2005). Cette voie consiste au passage du principe actif à
travers la partie gastro-intestinal (figure 28).

Cavité oral
Oesophage
Glandes salivaires
Foie
Vésicule biliaire
Estomac
Pancréas Intestin grêle
Gros intestin (Colon)
Rectum
Figure 28. Anatomie de la partie
gastro-intestinal humaine (F. H. Martini, 2002).
Une fois administré par la bouche, le principe actif
passe rapidement à la partie gastrointestinale. Le long de cette partie,
le principe actif est absorbé principalement au niveau de
l'iléon, mais sa biodisponibilité peut être réduite
à cause du premier passage hépatique. Ce dernier désigne
toute transformation du principe actif au niveau du foie. Enfin, le principe
actif passe dans la circulation générale à travers la
veine cave (J. Jean-Francois, 2004).
La partie gastro-intestinale est le site d'absorption
préféré pour divers agents thérapeutiques à
cause de la facilité d'administration et le confort aux patients. Les
formes galéniques orales consistent en des comprimés et des
capsules lesquelles sont souvent fourni comme des systèmes à
libération instantanée (V. V. Ranade a, 1991 ;
V. V. Ranade b, 1991). Cette partie est aussi
caractérisée par les différentes valeurs de pH comme le
montre le tableau 12.
Tableau 12. Physiologie de la partie
gastro-intestinale humaine (J. W. Fara, 1989).
|
Région
|
Longueur
(m)
|
Surface
(m2)
|
pH
|
Nombre de bactérie
(cfu/ml)
|
Temps de
résidence (h)
|
|
Bouche
|
0,20
|
0,10
|
1,0-2,5
|
102
|
0-2 (à vide)
|
|
Intestin grêle 3-5
|
|
duodénum
|
0,25
|
1,90
|
5,5-6,0
|
102
|
|
|
jéjunum
|
2,80
|
184
|
6,0-7,0
|
105
|
|
|
iléon
|
4,20
|
276
|
7,0-7,5
|
107
|
|
|
Gros intestin 20-30
|
|
cæcum
|
0,20
|
0,05
|
6,4-7,0
|
108
|
|
|
côlon
|
1,50
|
0,25
|
7,0-7,5
|
1012
|
|
L'administration de médicaments par voie orale
possède des avantages et aussi des inconvénients (A. M.
Hillery, 2005).
Les avantages sont :
- Acceptabilité et confort du patient,
- Voie orale présente une grande surface efficace à
l'absorption du principe actif (
ex. la surface de l'intestin grêle est
200 m2),
- Muqueuse gastro-intestinale possède une surface
vascularisée importante, ce qui assure une absorption rapide de
médicament,
- Possibilité de retenir le principe actif dans la partie
gastro-intestinal si un système de vectorisation approprié est
utilisé, par conséquent, la fréquence de la dose
diminue
- Voie orale offre un potentiel pour réaliser une
libération contrôlée d'ordre zéro,
- Coût de la thérapie orale est
généralement plus faible comparativement aux autres voies
d'administration.
Les inconvénients sont :
- Vitesse d'absorption du principe actif à partir des
formes galéniques conventionnelles est affectée par plusieurs
facteurs, à savoir fluctuation du pH dans l'estomac et l'intestin
grêle, présence ou absence de la nourriture, rythme
journalier...
- Possibilité d'avoir des effets secondaires, par exemple
certains principes actifs sont gastrotoxiques en causant des dommages dans la
muqueuse de l'estomac.
II.3.3. Voie nasale
La cavité nasale a été prévue
comme un site d'action pour certains principes actifs. Elle peut être
utilisé pour diriger directement le principe actif vers le
système circulatoire car le nez est très vascularisé
(A. M. Hillery, 2005).
Le nez est le site d'entré primaire de l'air dans le
système respiratoire et joue un rôle de filtration et
d'humidification de l'air inspiré. A l'intérieur, le nez donne
accès au nasopharinx et conduit à la tranchée et à
l'oesophage.
La cavité nasale est composée
d'épithélium squameux, respiratoire et olfactif. Les voies
d'administration proposées pour la cavité nasale sont de type
transcellulaire, paracellulaire. En effet, la voie transcellulaire s'effectue
à travers les cellules épithéliales par l'un des
mécanismes suivant : diffusion passive, transport par un vecteur et
processus endocytique. La voie paracellulaire s'effectue entre les cellules
épithéliales adjacentes avec un mécanisme de type
diffusion passive.
En dépit de sa facilité d'utilisation, la voie
nasale présente un certains nombre d'obstacles qui peuvent bloquer
l'absorption de principes actifs. Parmi ces obstacles citant, le mucus qui
tapisse l'épithélium de la cavité nasale qui est de nature
visqueuse et permet de piéger les particules inhalées, le poids
moléculaire du principe actif sachant que la vitesse d'absorption d'un
médicament est inversement proportionnelle à son poids
moléculaire (J. Jean-François, 2004 ; A. M. Hillery,
2005).
La voie nasale présente plusieurs avantages et aussi un
certains nombre d'inconvénients (A. M.
Hillery, 2005).
Les avantages sont :
- Cavité nasale présente une surface d'absorption
de principes actifs relativement grande (~ 100 cm2),
- Cavité nasale est très vascularisée ce qui
assure une absorption rapide de médicaments,
- Cavité nasale possède une activité
métabolique faible comparativement à la partie
gastrointestinale,
- Surface de la cavité nasale est facilement accessible
pour les vecteurs de médicaments, - Facilité lors de
l'administration.
Les inconvénients sont :
- Diffusion du principe actif peut être limitée par
la barrière physique de mucus,
- Pour les principes actifs de hauts poids moléculaires
(faiblement absorbés), la voie nasale est limitée seulement aux
molécules actives,
- Possibilité d'avoir des effets secondaires.
II.3.4. Voie pulmonaire
La voie pulmonaire permet une vectorisation de principes
actifs directement vers leur sites d'action ce qui réduit la
fréquence de la dose, tandis que la faible concentration du principe
actif, dans le système circulatoire, peut aussi réduire les
effets secondaires (A. M. Hillery, 2005).
La vectorisation par voie pulmonaire s'effectue de plusieurs
façons, aérosols, systèmes d'inhalation, poudres et
solutions (nébuliseurs) (T. Zecheru, 2008).
La vectorisation de médicaments par voie pulmonaire est
destinée au traitement des maladies respiratoires et elle semble de plus
en plus l'option viable pour la vectorisation
systémique de médicaments. Récemment, la
vectorisation des peptides et autres molécules qui
ne sont pas
absorbés dans la partie gastro-intestinale peut être
effectuées par voie pulmonaire
(T. Zecheru, 2008 ; A. M. Hillery, 2005).
La partie respiratoire chez l'homme est divisée en deux
voies aériennes, inférieures et supérieures. Les voies
aériennes supérieures contiennent le nez, pharynx, larynx,
tranchée et bronches. Par contre les voies aériennes
inférieures sont constituées de bronchioles terminales et des
alvéoles (J. Jean-François, 2004) (figure
29).

Sinus sphéroïde
Sinus frantal
Turbine moyenne Turbine inférieure Nasopharynx Oropharynx
Larynx
Tranchée Branche main
Branche subsegment large
Petit branche
Bronchiole
Bronchiole terminals
Bronchiole respiratoire Conduites et sacs alvéolaires
Voies aériennes pulmonaires/périphériques/
respiratoires
Voies aériennes supérieures
Voies aériennes centrales/conduite
Figure 29. Différentes régions
de la partie respiratoire humaine (A. M. Hillery, 2005).
Parmi les différents avantages de la voie pulmonaires
citant (A. M. Hillery, 2005) : - Dose nécessaire pour
avoir l'effet pharmacologique peut être réduit,
- Système de vectorisation agit rapidement à
travers cette voie,
- Formulation évite le premier passage
hépatique,
- Poumons offre une surface assez large pour l'absorption de
médicaments,
- Membranes des poumons sont plus perméables pour divers
composés comparativement à ceux de l'intestin grêle,
- Poumons présentent une surface vascularisée
importante, ce qui assure une absorption rapide de médicaments,
- Voie pulmonaire offre un environnement beaucoup moins agressif
pour de divers principes actifs (protéines peptides) comparativement
à la voie orale.
La voie pulmonaire possède aussi certains
inconvénients qui sont :
- Systèmes complexes sont nécessaires pour la
vectorisation de principes actifs vers les voies aériennes, mais ces
vecteurs peuvent être inefficaces,
- Systèmes aérosols peuvent être
difficilement utilisables,
- Absorption de médicaments peut être limitée
par la barrière physique du mucus.
II.4. Utilisation des polymères
biodégradables pour la libération contrôlée de
principes actifs
Dans un premier temps, la majorité des polymères
utilisés pour la vectorisation de principes actifs sont de nature
hydrophobes et nondégradables comme le poly(diméthylsiloxane),
polystyrène, polyuréthane...etc. Mais l'application de tels
polymères comme vecteurs de principes actifs est limité par la
nécessité d'une deuxième intervention chirurgicale pour
les extraire du corps. Pour y remédier, des polymères
biodégradables naturels et synthétiques sont devenus des
candidats très attractifs pour la vectorisation de médicaments
(T. Allen, 2004).
Dans les systèmes polymères, le principe actif
est incorporé dans une matrice polymère. La vitesse de
libération du principe actif à partir de tels systèmes
dépend de plusieurs paramètres, à savoir la nature de la
matrice polymère, la géométrie de la matrice, les
propriétés du principe actif, la quantité du principe
actif initialement incorporée dans la matrice polymère et
l'interaction polymère-principe actif. Le mécanisme qui
régi la libération du principe actif à partir de la
matrice polymère peut être contrôlé physiquement ou
chimiquement. Les systèmes physiquement contrôlés peuvent
être classifiés en systèmes à diffusion
contrôlée et en systèmes à solvant
contrôlé. Les systèmes chimiquement contrôlés
peuvent être obtenus par la dispersion du principe actif dans la matrice
polymère biodégradable ou par le développement des
systèmes complexes biodégradables de type
polymère-principe actif (S. Lakshmi, 2006).
II.4.1. Différents types de profiles de la
vectorisation de principes actifs
On distingue cinq profiles différents
schématisés dans la figure 30 (A. K. Bajpai, 2008 ; N. B.
Graham, 1992) :

Temps
Figure 30. Différents types de
profiles de libération de principes actifs : (profile I) la vitesse de
libération varie exponentiellement avec le temps, (profile II) vitesse
de libération constante produisant une cinétique d'ordre
zéro, (profile III) libération d'ordre zéro avec effet
retard considérable, (profile IV) libération pulsée avec
effet retard, (profile V) libérations multiples avec des effets retards
constants intermédiaires (A. K. Bajpai, 2008).
Profile I : on remarque que la
libération du principe actif est prolongée, mais avec une vitesse
qui n'est pas constante.
Profile II : dans ce cas, la
libération est constante ou d'ordre zéro. En effet, le principe
actif est libéré avec une vitesse constante de sorte que sa
concentration dans le sang est maintenue à un niveau
thérapeutique efficace.
Profile III : on remarque que la
libération du principe actif est considérablement
prolongée au début, puis devient constante. De tels
systèmes sont utilisés pour la vectorisation de principes actifs
pendant la nuit.
Profile IV : on remarque un
prolongement de la libération suivi par une libération brusque du
principe actif. Ce types de profiles tient compte aussi de la libération
de principes actifs pendant la nuit et aussi de la vectorisation des
hormones.
Profile V : dans ce cas, on remarque
plusieurs pulsations à des périodes spécifiques.
II.4.2. Différents systèmes utilisés
pour la vectorisation de principes actifs Selon le mécanisme de
la libération, on distingue :
II.4.2.1. Systèmes à diffusion
contrôlée
a/ Système réservoir (membrane)
C'est le système le plus simple. Il s'agit d'un noyau
de principe actif dissous, suspendu ou compressé entouré d'une
membrane polymère insoluble dans l'eau (figure 31). La
géométrie de ce type de systèmes est
généralement sphérique, cylindrique ou sous forme d'un
disque. Le mécanisme qui régisse la libération du principe
actif à travers la membrane est souvent de type solution-diffusion. En
effet, d'abord le principe actif se dissous dans l'un des côtés de
la membrane polymère (i.e. dispersion du principe actif dans la membrane
polymère), suivi de sa diffusion à travers cette membrane, enfin
le principe actif se désorbe à partir de l'autre
côté de la membrane (A. K. Bajpai, 2008 ; D. Jones, 2004 ;
V. V. Ranade, 2004).
Le système réservoir peut rencontrer certains
problèmes, par exemple dans le cas d'une rupture accidentelle dans la
membrane, une grande quantité du principe actif peut être
libérée dans le sang : ce phénomène est
appelé décharge du principe actif (V. V. Ranade,
2004).

Temps
Figure 31. Libération d'un principe
actif à partir d'un système réservoir : (a) système
implantable ou oral, (b)
système transdermale (L.
Brannon-Preppas, 1997).
b/ Système matriciel (monolithique)
Le système matriciel consiste en un principe actif
dissous ou dispersé d'une manière homogène dans toute la
matrice polymère (figure 32). La vitesse de libération du
principe actif à partir de ce type de systèmes est constante
(uniforme) (A. K. Bajpai, 2008 ; V. V. Ranade, 2004).
Il existe plusieurs approches pour réaliser le
système matriciel, citant ce qui suit (A. K.
Bajpai, 2008) :
i. Méthode de compression directe : il s'agit de
mélanger des particules de polymère et de principe actif suivi
par une compression directe pour avoir un comprimé.
ii. Dissolution du polymère et le principe actif dans un
solvant approprié (adéquat) suivi par une extraction du
solvant.
iii. Incorporation du principe actif dans un polymère
par polymérisation du mélange principe actif-monomère ou
par le gonflement d'un hydrogel dans une solution du principe actif.

Temps
Figure 32. Libération d'un principe
actif à partir d'un système matriciel à libération
contrôlée (L. Brannon-
Preppas,
1997).
II.4.2.2. Systèmes chimiquement
contrôlés
Dans ce type de systèmes, la libération du principe
actif s'effectue, en général, dans un milieu aqueux par un ou
plus des mécanismes suivants :
i. Biodégradation progressive du système
polymère contenant le principe actif ;
ii. Biodégradation de la liaison instable qui lie le
principe actif au polymère ;
iii. Diffusion du principe actif à partir du
système polymère.
a/ Systèmes bioérodibles et
biodégradables
Dans ce cas, le principe actif est incorporé dans une
matrice polymère biodégradable. La libération du principe
actif implique la décomposition hydrolytique ou enzymatique
progressive du polymère de telle sorte que cette
décomposition peut être superficielle ou volumique (V. V.
Ranade, 2004).
Les systèmes biodégradables sont avantageux, car
d'une part leur utilisation élimine la nécessité d'une
deuxième intervention chirurgicale pour les extraire du corps et d'autre
part pour leurs petites tailles et leur faible coût. En plus, les
matériaux biodégradables ainsi que leurs métabolites
doivent être non-toxiques, non-cancérigènes et non
tératologiques (A. K. Bajpai, 2008). La figure 33
explique le mécanisme de la libération d'un principe actif
à partir d'une matrice polymère biodégradable.
|
Matrice polymère
|
|
Particules du principe actif
|
Figure 33. Libération du principe actif
à partir des systèmes biodégradables : (a) érosion
volumique, (b) érosion
superficielle (L. Brannon-Preppas,
1997).
b/ Systèmes à chaînes
pendants
Dans ce type de systèmes, les molécules du
principe actif sont liées au squelette du polymère par des
liaisons chimiques soit directement ou par l'intermédiaire d'une autre
molécule qui serve de lien (figure 34). La décomposition
enzymatique ou hydrolytique de ces liaisons provoque la libération du
principe actif avec une vitesse contrôlée. Les systèmes
à chaînes pendants sont utilisés pour une libération
contrôlée et ciblée, i.e. ils peuvent être
destinés vers un type de cellules ou tissues spécifiques
(A. K. Bajpai, 2008 ; V. V. Ranade, 2004).
Squelette du polymère

Principe actif
Eau ou Enzymes
Temps 0 Temps t
Figure 34. Clivage de squelette
polymère (T. J. Kreeger, 1993).
II.4.2.3. Systèmes à solvants
activés On distingue deux types de systèmes :
a/ Système à gonflement
contrôlé
La libération du principe actif dans certains
systèmes s'effectue au fur et à mesure que la matrice
polymère se gonfle (i.e. changement dans les dimensions de la matrice
polymère (V. V. Ranade, 2004) (figure 35). L'exemple
typique de cette catégorie de systèmes est l'hydrogel. Ce dernier
consiste à une structure enchevêtrée constituée de
chaînes macromoléculaires réticulées. Lorsque la
matrice hydrogel se trouve au contact d'un solvant thermodynamiquement
compatible, les chaînes polymères se relaxes (S. Shukla,
2003). Ce phénomène est lié à la
température de transition vitreuse du polymère, par
conséquent, on distingue deux possibilités :
i. Si la température de transition vitreuse
(Tg) de polymère est inférieure à la
température expérimentale, on dit que le polymère est dans
son état caoutchouteux et par conséquent ses chaînes
deviennent plus mobiles et permettent une pénétration facile du
solvant dans l'hydrogel chargé du principe actif et une
libération ultérieure de ce dernier (A. K. Bajpai, 2008 ;
R. A. Grinsted, 1992).
ii. Si la température expérimentale est
inférieure à Tg de polymère, les chaînes
polymères dans l'hydrogel ne sont pas suffisamment mobiles pour
permettre une pénétration immédiate du solvant (A.
K. Bapjai, 2008).

Système réservoir gonflé
Système matriciel gonflé
Libération du
principe actif
Figure 35. Libération du principe actif
à partir des systèmes à gonflement contrôlé :
(a) système réservoir, (b)
système matriciel
(T. J. Kreeger, 1993).
b/ Systèmes osmotiquement
contrôlés
Dans le cas des systèmes osmotiquement
contrôlés, la libération du principe actif s'effectue sous
l'effet d'une différence de pression entre le système et le
milieu biologique environnant. Les premiers systèmes osmotiques ont
été développés par Alza Corporation dans les
années 70.
Les systèmes osmotiquement contrôlés sont
des pompes osmotiques constituées d'un noyau (ou réservoir) de
principe actif entouré par une enveloppe polymère
semi-perméable osmotiquement active. Le contact de ces systèmes
avec le fluide biologique permet à ce dernier de pénétrer
à l'intérieur du réservoir à travers la membrane
semi-perméable, ce qui provoque la dissolution du principe dans le
fluide biologique pour former une solution saturée dans le noyau. Le
nombre de molécules de principe actif qui passent en solution est
proportionnelle à la pression osmotique dans le noyau. Comme la membrane
semi-perméable est rigide, donc pour réduire la pression à
l'intérieur du système, la solution saturée du principe
actif doit quitter le noyau vers le milieu environnant à travers un
petit orifice (f. Jones, 2004 ; V. V. Ranade, 2004). La figure
36 résume le mécanisme général qui régi ce
genre de systèmes.

Principe actif expulsé
Orifice
Eau pénétrée

Noyau de principe actif
Membrane semi-perméable
Figure 36. Pompe osmotique (T. J.
Kreeger, 1993).
II.4.3. Différents types de vecteurs de principes
actifs et leurs architectures II.4.3.1. Architecture des
polymères
L'architecture du polymère décrit la forme d'une
seule molécule du polymère qui détermine souvent ses
propriétés physicochimique (L. Y. Qiu, 2006). Il
existe plusieurs architectures des polymères qui sont :
a/ Polymère linéaire
Le polymère linéaire possède la forme
architecturale la plus simple. L'application typique des polymères
linéaires se concentre dans la fabrication des conjugués
polymère-principe actif. Les années 50 marquent la naissance de
la première proposition pour relier un principe actif à une
chaîne polymère. Le modèle général pour
mettre en évidence le conjugué polymère-principe actif
à été proposé par Ringsdorf (H. Rinsdorf,
1975). Le modèle est basé sur la combinaison de la
chimie et la biologie. Il contient les éléments suivants :
squelette polymère, principe actif, lien, groupement de ciblage et
groupement de solubilisation. La figure 37 représente un schéma
explicatif de ce modèle.
Squelette du polymère

Lien
Groupement de ciblage
Principe actif
Groupement de solubilisation
Figure 37. Modèle de Ringsdorf pour le
conjugué polymère-principe actif (H. Rinsdorf,
1975).
Les différentes formes architecturales des
polymères linéaires utilisées dans le domaine de la
vectorisation de principes actifs sont présentées dans la figure
38 suivante.

Homopolymère Copolymère diblock type AB
Copolymère triblock type ABA

Copolymère aléatoire

Copolymère triblock type BAB

Copolymère alterné
Copolymère triblock type ABC
Figure 38. Différentes formes
architecturales des polymères linéaires (L. Y. Qiu,
2006).
b/ Polymères branchés
Des efforts ont été effectués pour
comprendre le mécanisme de polymérisation de la synthèse
des polymères branchés (L. Y. Qiu, 2006). Selon
les propriétés du squelette polymère et l'embranchement,
deux méthodes générales ont été
appliquées pour la synthèse des polymères greffés.
La première méthode consiste à une copolymérisation
directe de deux monomères ou plus. Tandis que la deuxième
méthode consiste à une utilisation d'un squelette polymère
en présence des sites actifs polyfonctionnels comme initiateurs
(H. Q. Wie, 1999 ; Y. X. Li, 1997). Les polymères
étoiles sont des structures tridimensionnelles où des bras de
mêmes masses moléculaires ou de masses différentes
émanent d'un noyau central (M.-C. Jones, 2003). Parmi
les polymères branchés, on trouve aussi les dendrimères.
Ces derniers représentent une nouvelle classe de macromolécules
possédant une structure tridimensionnelle dans laquelle une série
de branches se développent à partir d'un noyau central
(F. Aulenta, 2003 ; E. R. Gillies, 2005). Un dendrimère
typique contient trois parties principales : noyau central multifonctionnels,
unités embranchées, et groupements superficiels. Les
différentes formes
architecturales des polymères branchés sont
schématisées dans la figure 39 suivante (L. Y. Qiu,
2006).
|
|
Polymère en forme d'étoile
|
Copolymère block en forme d'étoile
|
Block dendrimère Copolymère dendri-griffé


Copolymère griffé

Dendrimère

Polymère hyperbranché
Figure 39. Différentes formes
architecturales des polymères branchés (L. Y. Qiu,
2006).
c/ Polymères réticulés
Il existe plusieurs formes architecturales des polymères
réticulés, qui sont schématisés dans la figure 40
ci-dessous.

Réseau polymère Réseaux polymères
interpénétrés
Réseaux polymères
semi-interpénétrés
Figure 40. Différentes formes
architecturales des polymères réticulés (L. Y.
Qiu, 2006).
II.4.3.2. Différents types de vecteurs de principes
actifs
La structure des vecteurs polymères chargés en
principe actif dépend du procédé de fabrication
utilisé, de la nature des matériaux polymères et le
caractère lipo- ou hydrosoluble du principe actif (A.
Wawrezinieck, 2008).
Les premières études pour la conception des
vecteurs pharmaceutiques à base de polymères
biodégradables ont été basées sur des formulations
monolithiques. Ces dernières consistent à des matrices
bidimensionnelles d'une géométrie adéquate. Cependant, les
matrices monolithiques possèdent certains problèmes
intrinsèques, à savoir la nécessité d'être
implantés à l'intérieur du corps et aussi leurs faibles
vitesses de libération de principes actifs, en particulier dans le cas
des principes actifs faiblement perméables comme dans le cas des
stéroïdes (S. Lakshmi, 2006). Pour
améliorer les vitesses de libération, de nouvelles formes ont
été utilisées tel que des microsphères et des
microcapsules. Les microsphères sont des formes monolithiques où
le principe actif est distribué dans une matrice polymère alors
que les microcapsules consistent à un principe actif encapsulé
dans une coquille polymère (E.
Mathiowitz, 1999 ; R. Arshady, 1999).
Les liposomes sont des vecteurs de médicaments
très attractifs car ils sont facilement administrés par voie
intraveineuse d'une part et ils sont similaires aux membranes cellulaires
d'autre part. Cependant, la faible stabilité des liposomes a
poussé le développement de nouvelles formes galéniques
à base de polymères qui sont des nanosphères et
nanocapsules de diamètre inférieur à un micromètre
(S. Lakshmi, 2006).
L'hydrogel forme un autre type de vecteurs matriciels pour la
vectorisation de principes actifs, en particulier dans le cas des
protéines et des peptides de hauts poids moléculaires
(N.
A. Peppasa, 2000).
Tous les systèmes décrits
précédemment sont destinés à la vectorisation
contrôlée de principes actifs d'une part, et aussi pour maintenir
la concentration en principes actifs constante dans le corps sur une longue
période. Cependant, ces systèmes ne sont pas adaptés pour
la vectorisation de tout type de principes actifs. Par conséquent, des
systèmes de vectorisation pulsé ont été
développés pour la libération d'une certaine
quantité du principe actif pendant une courte période
après un déphasage de temps. On distingue la vectorisation
pulsée intelligente où la libération de principes actifs
est contrôlée par des stimuli internes ou externes, et la
vectorisation programmable où le principe actif est automatiquement
libéré à partir de la matrice à un temps
prédéterminé (L. Y. Qiu, 2001). La figure
41 ci-dessous résume les différentes formes de vecteurs
pharmaceutiques.

Polymères dendritiques multifonctionnels



Vecteurs Pharmaceutiques
Critaux liquide Nanosphères
Nanocapsules
Figure 41. Vecteurs pharmaceutiques
(C. Kaprarissides, 2006).
II.4.3.2.1. Micelles polymériques
Les micelles sont des nanovecteurs idéals pour les
agents insolubles. Ils sont constitués de copolymères qui
consistent à des unités monomériques hydrophiliques et
hydrophobiques. La formation des micelles polymériques résulte de
la diminution de l'énergie libre obtenu par l'association des
unités hydrophobes entre elles afin de réduire le contacte avec
le milieu aqueux environnant (J. Jean-François, 2004 ; V. P.
Torchilin, 2004 ; N. A. Pappasa, 2000 ; V. P. Torchilin, 2001).
L'auto-association des copolymères amphiphiles dans un
milieu aqueux mène à la formation des micelles sphériques
de tel sorte que l'extrémité hydrophile du copolymère est
orientée vers la surface de la micelle tandis que
l'extrémité hydrophobe est orientée vers le noyau de la
micelle (N. Hasirci, 2007). En général, lorsque
le segment hydrophobique du block amphiphile est plus long que le segment
hydrophilique, la micelle aura une forme sphérique. Dans le cas
contraire, les blocks copolymères forment des structures
non-micellaires, à savoir des tiges, et des lamelles (V. P.
Torchilin, 2004).
Il existe au moins trois catégories principales de
micelles à base de blocks copolymères linéaires
(L. Y. Qiu, 2006) :
- Micelles de blocks copolymères communs,
- Micelles de conjugué copolymère-principe
actif,
- Micelles de complexes ionomères.
La figure 42 illustre les différentes catégories de
micelles.

Block hydrophilique
Block Principe actif
hydrophobique
Figure 42. Trois principaux types de micelles
à base de block copolymère linéaire : (a) micelle de
block
copolymère commun, (b) micelle de conjugué block
copolymère-principe actif, (c) micelle de complexe
block
ionomère (L. Y. Qiu, 2006).
Les micelles polymériques sont thermodynamiquement
stables dans le milieu physiologique, à cause de leur faible
concentration micellaire critique. Le diamètre des micelles ne
dépasse pas, généralement, 100 nm. Ceci leur donne la
possibilité de circuler à long terme dans le sang et
améliore leur perméabilité dans les cellules
endothéliales à proximité des tumeurs par diffusion
passive. La vectorisation ciblée des micelles peut être
améliorée si des ligands ou des anticorps sont fixés sur
sa surface (N. Hasirci, 2007).
Le noyau des micelles polymériques pharmaceutiques doit
montrer une capacité de chargement élevée, un profile de
libération contrôlé de principe actif et une bonne
compatibilité avec le principe actif incorporé (V. P.
Torchilin, 2004).
L'incorporation du principe actif dans la micelle
polymérique dépend des propriétés physicochimiques
du block copolymère. La première procédure consiste
à une dissolution directe du block copolymère avec le principe
actif dans un solvant aqueux. Cette procédure est plus utilisée
dans le cas de copolymères modérément hydrophobiques, et
peut exiger le chauffage de la solution aqueuse pour provoquer la formation de
micelles par la déshydratation du segment formant le noyau. La
deuxième procédure fait appel à des copolymères
amphiphiles peu solubles dans l'eau et à un solvant commun pour le
principe actif et le copolymère. Le mécanisme de formation des
micelles dans ce cas dépend de la procédure d'extraction du
solvant. En effet, si le solvant et l'eau sont miscibles, le mélange
copolymère peut être filtré par dialyse où
l'extraction lente de la phase organique provoque la formation des micelles. La
méthode solution-casting nécessite l'évaporation de la
phase organique pour aboutir à des films polymériques où
l'interaction polymère-principe actif est favorisée. La
réhydratation de ces films par le solvant aqueux chaud produit des
micelles chargées en principe actif. Si le solvant et l'eau ne sont pas
miscibles, l'incorporation physique du principe actif hydrophobe peut
être effectuée par le processus d'émulsion huile-
dans-eau (O/W). La figure 43 illustre les méthodes
d'incorporation du principe actif (G.
Gaucher, 2005 ; X. Shuai, 2004).

Figure 43. Procédures de chargement de
principes actifs : (A) équilibre simple, (B) dialyse, (C)
émulsion o/w,
(D) solution casting, et (E) lyophilisation (G.
Gaucher, 2009).
II.4.3.2.2. Vésicules polymériques (ou
polymèrsomes)
En fonction de leurs architectures et des conditions de
préparation, des blocks copolymères amphiphiles peuvent
s'auto-associent pour former différents agrégats, à savoir
des sphères, tiges, micelles tubulaires, lamelles, ou des
vésicules. Des vésicules polymériques possèdent une
structure similaire à celle des liposomes, où une cavité
hydrophile est entourée par une membrane polymérique hydrophobe
appelées polymèrsomes (L. Y. Qiu, 2006 ; H. Kakula,
2002).
Les polymèrsomes présentent certains avantages
comparativement aux liposomes, en plus de leur stabilité, ils ont la
possibilité de réguler l'épaisseur de leur membrane
(L. Y. Qiu, 2006).
La méthode la plus utilisée pour la
préparation des vésicules géantes avec des
diamètres de l'ordre de 10 à 200 um est l'electroformation. Dans
ce processus, un courant alternatif est appliqué sur un film
polymérique dans une solution aqueuse. La taille des vésicules
peut être facilement contrôlée par la variation de voltage
et de la fréquence du champ électrique (L. Y.
Qiu, 2006).
Une autre méthode appelée injection est
appliquée. La solution de polymère organique est injectée
lentement dans une solution aqueuse pour précipiter le
polymèrsome (F. Meng, 2003). Souvent, l'addition
directe de l'eau dans une solution de polymère organique est aussi une
autre méthode utilisée pour la préparation des
polymèrsomes (B. M. Disher, 2000).
La formation des vésicules polymériques à
partir des copolymères di- et triblock est illustrée dans la
figure 44 suivante.

Copolymère diblock
Copolymère triblock
Figure 44. Formation des vésicules
à base des copolymères di- et triblock (T. Zecheru,
2008).
II.4.3.2.3. Micro et nanoparticules
Dans la vectorisation des agents bioactifs,
généralement, l'agent actif est dissous, piégé,
adsorbé, attaché ou encapsulé dans une matrice
polymérique qui a des dimensions de l'ordre de micro ou
nanomètre. Selon la méthode de préparation, on distingue
des micro ou nanoparticules, sphères ou capsules de
propriétés et caractéristiques de libération
différentes. Les capsules sont des systèmes vésiculaires
où le principe actif est piégé dans une cavité
centrale entouré par une membrane polymérique, par contre les
sphères sont des systèmes où le principe actif est
dispersé physiquement d'une manière uniforme dans la matrice
(N. Hasirci, 2007). La figure 45 montre les deux types de
micro et nanoparticules.
Micro ou nanosphère (système monolithique)

Médicament Médicament
encapsulé Micro ou nanocapsule (système
réservoir) adsorbé
Figure 45. Les deux modèles de particules
utilisées en libération contrôlée (J.
Jean-Francois, 2004).
A cause de leurs propriétés physicochimiques
attractives, à savoir la taille, le potentiel superficiel et la balance
hydrophile/hydrophobe, les nano et microparticules ont été
développées au début des années 1970 comme vecteurs
utilisés pour la vaccination et pour le transport de médicaments
anticancéreux (J. Jean-Francois, 2004 ; A. K. Bajpai,
2008).
Les nanoparticules sont de formes colloïdales,
assimilées à des substances macromoléculaires de
diamètre variant entre 10 et 100 um. Lorsque le diamètre
dépasse le micron, soit entre 2 et 300 um on parle alors de
microparticules (A. K. Bajpai, 2008 ; E. Allémann,
1993).
Ils existent plusieurs méthodes utilisées pour
la préparation des micros et nanoparticules dont les principales sont la
polymérisation de monomères ou la dispersion de polymères
préformés. Par exemple, la technique d'émulsification est
utilisée pour la préparation des nanocapsules et des
nanosphères de poly(acide lactique) (C. Vautheir-Holtzsherer,
1991). Le tableau 13 suivant résume ces différentes
méthodes de synthèse.
Tableau 13. Méthodes de synthèse
des différents types de nano et microparticules (C.
Vautheir-Holtzsherer, 1991).
|
Type de particules
|
Méthodes de préparation
|
|
Microcapsules
|
- Coacevation
- Polymérisation interfaciale
|
|
Microsphères
|
- Evaporation du solvant
- Fusion à chaud
- Dénaturation chimique
- Réticulation chimique
|
|
Nanocapsules
|
- Emulsion-Polymérisation
- Emulsification
|
|
Nanosphères
|
- Emulsion-Polymérisation
- Emulsification
- Désolvatation
|
II.4.3.2.4. Hydrogels
Les hydrogels sont des réseaux tridimensionnels de haut
poids moléculaire composés d'un squelette polymérique, de
l'eau et d'un agent de réticulation. Ils sont très
utilisés dans le domaine médical et pharmaceutique, par exemple
comme organes artificiels et comme systèmes de vectorisation de
principes actifs (A. K. Bajpai, 2008 ; M. T. Razzak, 2001).
Les hydrogels sont des matériaux polymériques
(polymère naturels ou synthétiques) insolubles dans l'eau
à des températures et des pH physiologiques. Ils sont capable de
retenir une grande quantité d'eau ou de fluide biologique (> 20 %),
ce phénomène est appelé gonflement ou changement de
volume. Ce dernier peut être provoqué par un changement dans les
conditions externes comme pH, température, force ionique et courant
électrique (S. Lakshmi, 2006 ; A. K. Bajpai, 2008)
comme le montre la figure 46 suivante.

Changement dans la température, pH, force ionique etc.
Figure 46. Libération de principes
actifs à partir d'hydrogels sensibles aux stimuli et intelligents
(A. M. Hillery,
2005).
Les hydrogels peuvent être obtenus à partir des
homo- ou copolymères par réticulation physique ou chimique. La
réticulation physique est réalisée entre les
polymères par des interactions de type Van der Waals et la
réticulation chimique se fait entre les chaînes de
polymères par des liaisons covalentes ou des liaisons hydrogènes
(S. Lakshmi, 2006 ; K. R. Kamath, 1993).
Les systèmes de vectorisation à base d'hydrogels
présentent certains avantages, à savoir biocompatibilité
élevée, adaptation facile de la libération de principes
actifs par le contrôle du degré de réticulation ainsi que
le changement dans la structure de polymère qui forme l'hydrogel qui
protège le principe actif de la dégradation et de l'inactivation.
L'un des avantages significatifs des systèmes de vectorisation de
principes actifs à base d'hydrogels est leurs gonflements dans un milieu
aqueux. Ces matrices sont sensibles à des stimuli aussi variés
que le pH, température, force ionique et courant électrique. De
ce fait, deux types d'hydrogels sont à distingués : hydrogels
conventionnels qui sont conçus pour libérer leur contenu
localement sans stimulus externe, à un endroit
prédéterminé d'où la nécessité
d'utiliser des polymères biodégradables, et des hydrogels dits
intelligents qui serait, par contre, capable de libérer son principe
actif seulement lorsqu'un stimulus spécifique est
présent (J. Jean-Francois, 2004 ; S. Lakshmi,
2006).
II.5. Application de poly(acide lactique) dans le domaine
pharmaceutique
Le poly(acide lactique) est un polyester thermoplastique
biodégradable qui possède de bonnes propriétés
mécaniques dont les produits de dégradation sont non toxiques.
Par conséquent, le PLA est utilisé dans de nombreuses
applications (A. P. Gupta, 2007).
À cause de ses propriétés
mécaniques comparables aux autres polymères thermoplastiques
comme PS et PET, le poly(acide lactique) est utilisé pour la fabrication
de fibres, de films, de textiles et d'emballages thermoformées. Dans le
domaine de l'emballage, le PLA est utilisé pour la fabrication des sacs
de déchets, plats pour fruits et légumes, plats et tasses
jetables ... Dans le domaine de textiles, il est utilisé pour la
fabrication des rideaux, serviettes, habillements ... (A. P. Gupta,
2007 ; K. J. Jem, 2010).
Comme le PLA est plus léger et plus résistant
que les polymères conventionnels, des composites à base de PLA
développés par la firme Toyota sont utilisés dans le
domaine de l'automobile. Dans le domaine de l'électronique, le PLA est
utilisé pour la fabrication de PC portables, de disques compacts et des
bobines de films. Le poly(acide lactique) est aussi utilisé dans le
domaine de la construction à cause de sa faible inflammabilité en
cas d'incendie et de ses propriétés antibactériennes ainsi
que sa résistance aux rayonnements UV. Par exemple, il est
utilisé pour les capitonnages, les dalles de moquette et pour la
fabrication des stores (H. N. Rabetafika, 2006).
Comme le poly(acide lactique) et ses dérivés
sont bien tolérés et ne présentent aucune toxicité
vis-à-vis de l'organisme, on trouve qu'ils sont très
utilisés dans le domaine médical, à savoir fil de suture,
implants médicaux et en particulier dans le domaine de la vectorisation
de principes actifs (A. P. Gupta, 2007) ; H. N. Rabetafika,
2006).
En 1981, Gurny et al. (R. Gurny, 1981) ont
préparé des nanosphères de poly(D,L-acide lactique)
chargées en testostérone par la technique
d'évaporation/émulsion. Depuis la dernière
décennie, des microsphères à base de PLA sont obtenues et
employées dans l'immobilisation d'enzymes (T. Hayashi,
1991), comme vecteurs de libération d'agents
anticancéreux (L. Ilium, 2000) ou de peptides
thérapeutiques comme le goserline utilisée pour le traitement de
l'endométriose (K. Nishimura, 1986).
La figure 47 suivante résume les différents
domaines d'application de PLA.

Sutures
Ingénierie tissulaire
Textiles
Emballage Pharmacie
Compost
Poly(acide lactique)
Vectorisation de princips actifs
Orthopédie
Figure 47. Application de poly(acide lactique)
(A. P. Gupta, 2007).
II.5.1. Rôle de l'interaction
polymère/principe actif dans la libération prolongée de
principes actifs
La libération du principe actif à partir d'une
matrice polymère est régi par trois mécanismes : (a)
diffusion Fickienne à travers la matrice polymérique, (b)
diffusion à travers des pores remplis d'eau crées lors de
gonflement de la matrice polymérique et (c) libération lors de
l'érosion de la matrice polymérique. Cependant, il a
été montré que l'interaction polymère-principe
actif joue un rôle significatif sur la libération de principe
actif (C. S. Proikakis, 2006). En effet, divers agents ont
été liés aux différents systèmes
polymériques au moyen des liaisons dégradables. Ces
systèmes polymériques sont hydrolysés dans le corps vivant
en libérant le principe actif à une vitesse
prédéterminée. Des polymères fonctionnels contenant
des agents bioactifs ont été préparés, soit par une
modification chimique de polymères préformés, ou par une
copolymérisation directe de monomère fonctionnel souhaité
avec le principe actif adéquat (M. Babazadeh, 2006).
L'interaction polymère-principe actif peut être de
type covalente, électrostatique ou par pont hydrogène.
Mirzaagha Babazadeh (M. Babazadeh, 2006) a
essayé de synthétiser un complexe polymère acrylique-
ibuprofène par la méthode de copolymérisation entre le
monomère acrylique et la molécule d'ibuprofène. Il a
estimé que la molécule d'ibuprofène peut être
liée à la chaîne du polymère acrylique au moyen
d'une liaison ester clivable au contact de milieu physiologique comme le montre
le schéma 2 suivant.
X
* H2C C CH2C
*



H
O
CH2C CH2 O C
Y
Principe actif
C



Schéma 2. Représentation de la liaison ester entre
la chaîne polymère et le principe actif (Acrylique/IB).
Bingbing Jiang et al. (B. Jiang, 2005) ont
préparé des nanoparticules à partir d'un polymère
chargé positivement qui est DEAE-dextran et d'un principe actif qui est
l'ibuprofène par la méthode de co-précipitation. Les
nanoparticules ont été formées avec succès à
cause d'une interaction électrostatique entre DEAE-dextran chargé
positivement et les molécules d'ibuprofène qui sont
chargées négativement, comme il est montré dans le
schéma 3 suivant.
pH élévé pH faible

DEAE-dextran Ibuprofène
Schéma 3. Possibilité d'une
liaison électrostatique entre le polymère et le principe actif
(DEAE/IB).
S. G. Kazarian et G. G. Martirosyan (S. G. Kazarian,
2002) ont étudié l'imprégnation de
l'ibuprofène dans le polyvinylpyrrolidone (PVP) par les méthodes
ATR-IR et Raman. Ils ont constaté qu'il ya une interaction entre le PVP
et les molécules d'ibuprofène. Cette interaction est de type par
pont hydrogène entre le groupe hydroxyle de l'ibuprofène et le
groupe carboxyle de PVP comme le montre le schéma 4 suivant.

HO
N
O
O
H2 C
H
C
Schéma 4. Possibilité d'une
liaison hydrogène entre le polymère et le principe actif
(PVP/IB).
II.5.2. Quelques exemples d'application de poly(D,L-acide
lactique) dans le domaine de la vectorisation de principes actifs
II.5.2.1. Chimiothérapie du cancer
Le cancer est une maladie qui a pour cause la division et la
prolifération cellulaires incontrôlées. Ce
phénomène est causé par plusieurs facteurs, certains
d'entre eux sont exogènes (maladies infectieuses, produits chimiques,
tabagisme, exposition aux radiations) et d'autres sont endogènes (profil
génétique, hormones). Le cancer est considéré comme
l'une des premières causes de mortalité chez l'homme, avec plus
de 150 000 nouveaux cas répertoriées au canada en 2006.
Parmi les méthodes traditionnelles utilisées
pour le traitement du cancer on trouve la chirurgie. Cette dernière a
été utilisée, particulièrement, pour les tumeurs
solides et pour éliminer les foyers tumoraux visibles. Cependant, les
foyers de cellules métastatiques difficilement perçus ne peuvent
être retirés par une intervention chirurgicale. Par
conséquent,
d'autres méthodes à caractère traditionnel,
sont souvent envisagées, à savoir la radiothérapie, la
chimiothérapie et l'immunothérapie (B. A. Chabner,
2006).
La chimiothérapie consiste à un traitement
médical ayant recours à de substances chimiques. Cependant,
à la différence d'autres classes d'agents chimiques, les agents
anticancéreux engendrent une toxicité importante dans
l'organisme. Malgré son développement au fil des années,
la chimiothérapie se trouve limitée par plusieurs obstacles
à savoir les mécanismes de résistance
développés par les cellules tumorales vis-à-vis des
différents types d'agents anticancéreux et leurs
mécanismes d'action. Parmi ces mécanismes, on trouve (D.
R. Ryan, 2006) :
i. Altération de la cible intracellulaire du
médicament ainsi qu'une diminution des voies d'activation
anticancéreux au niveau cellulaire.
ii. Plusieurs médicaments anticancéreux sont
labiles et susceptibles d'être dégradés au contact du
milieu physiologique.
De plus, les traitements chimiothérapeutiques
génèrent une toxicité importante dans l'organisme. En
effet, cette toxicité est attribuable en partie au manque de
spécificité des agents anticancéreux, ce qui provoque une
distribution de médicaments non spécifique dans l'organisme
affectant les tissus sains et n'atteignant qu'en faible quantité les
tissus tumoraux (J. L. Grem, 2006) (figure 48A). D'autre part,
plusieurs agents anticancéreux puissants possèdent une
solubilité aqueuse limitée, et malgré leur dissolution
dans des solvants organiques, il ya risque de précipitation au site
d'injection suite à la diffusion rapide du solvant dans le sang
(G. Gaucher, 2009).
Plusieurs molécules chimiques sont utilisées
pour le traitement du cancer, citant à titre d'exemple paclitaxel (PTX),
doxorubicine, étoposide, alcaloïde de la vinca, topotécan
(G. Gaucher, 2009) et bléomycine (T.
Kumanohoso, 1997).

Figure 48. Les stratégies de ciblage
d'un principe actif (PA) anticancéreux. (A) libération du PA tant
au niveau
des tissus tumoraux que les tissus sains, (B) libération du
PA dans la tumeur et internalisation du PA par les
cellules tumorales, (C)
internalisation du vecteur suite à l'interaction spécifique entre
la molécule de
reconnaissance attachée à la surface du
vecteur et son récepteur sur la membrane des cellules cibles,
(D)
libération du PA en réponse à l'environnement ou
à un stimulus externe (T. Lammers, 2008).
Dans le but d'avoir une formulation de principe actif qui
serait mieux tolérée, plusieurs types de systèmes
colloïdaux à base de polymères ont été
appliqués. Pour le ciblage d'un anticancéreux au niveau de la
tumeur solide, le vecteur idéal doit avoir un temps de circulation
plasmatique prolongée. Ensuite, le vecteur doit sortir de la circulation
sanguine et
s'accumuler dans le tissu tumoral. Enfin, une fois dans le
tissu cible, le vecteur doit libérer son contenu pour que le principe
actif puisse agir au niveau de sa cible intra ou extracellulaire (figures 46B,
46C et 46D). Parmi les polymères utilisés, on trouve les
polyesters synthétiques, en particulier, le poly(acide lactique) et ses
dérivés (G. Gaucher, 2009).
Le poly(acide lactique) est un polyester aliphatique ayant
trois configurations stéréochimiques qui sont poly(D-acide
lactique) (PDLA), poly(L-acide lactique) (ou PLLA) et le poly(D,L-acide
lactique) (ou PDLLA). Ce dernier est très utilisé comme vecteur
de principes actifs soit sous forme d'un homopolymère ou
copolymère (ex. PLGA).
II.5.2.1.1. Utilisation du PDLLA pour améliorer
l'efficacité thérapeutique de l'agent anticancéreux
bléomycine (BLM)
La bléomycine est un agent anticancéreux soluble
dans l'eau utilisé pour le traitement de nombreux cancers. Cependant,
cet agent est rapidement absorbée dans le sang avec une faible
distribution sélective, ce qui provoque une toxicité importante
dans l'organisme. Pour réduire la toxicité et maintenir la
concentration de la bléomycine élevée dans les
lésions, divers systèmes de chimiothérapie locale ont
été développés (M. Shimada,
1993).
Parmi ces systèmes, citant celui à base de
poly(D,L-acide lactique). Toru Kumanohoso et al. (T. Kumanohoso, 1997)
ont préparé un nouveau système de vectorisation
de la bléomycine pour le ciblage des lésions. Ce système
est préparé par l'incorporation de la bléomycine dans un
petit cylindre de PDLLA de faible masse moléculaire (Mw =
3500 Da) par la technique de pressurage à l'état fondu.
Dans le but de déterminer l'efficacité de la
nouvelle formulation BLM-PDLLA comparativement à la solution de PLM
(BLM-SOL), un test in vitro (pH = 7,4 à 37 °C) pour l'étude
de la libération de PLM à partir de la formulation BLM-PDLLA a
été effectué d'une part, et d'autre part, des
expériences in vivo sur des rats ont été aussi
réalisées. Toru Kumanohoso et al. (T. Kumanohoso, 1997)
ont choisi 84 rats divisés en deux groupes pour tester les deux
formulations, la formulation BLM-SOL est injectée dans le dos de chaque
rat du groupe 1 (48 rats), et la formulation BLM-PDLLA est implantée
dans le dos de chaque rat du groupe 2 (36 rats).
Les résultats trouvés pour le test in vitro sont
montrés sur la figure 49. D'après cette dernière, le
cylindre BLM-PDLLA se dissous progressivement au fil du temps jusqu'à sa
disparition complète après 35 jours. Cela signifie que BLM est
libéré à partir de la formulation BLM-PDLLA lentement sur
une période dépassant les trois semaines.

Temps (jours)
Figure 49. Libération de la
bléomycine à partir de BLM-PDLLA dans le milieu
physiologique
(pH = 7,4 à 37°C).
Pour le test in vivo, l'activité de BLM au cours du
temps a été mesurée dans 2 cm de tissu sous-cutané
autour du site d'implantation dans le cas des cylindres BLM-PDLLA et du site
d'injection dans le cas de la solution BLM-SOL. Une activité
élevée a été maintenue dans le tissu
sous-cutané dans le cas des implants BLM-PDLLA pour au moins 14 jours.
Par contre, la solution BLM-SOL n'a démontré aucune
activité au-delà de 3 jours (figure 50).

BLM-PDLLA
BLM-SOL
Temps (jours)
Figure 50. Variation de la concentration de
BLM dans les tissus sous-cutanés autour de site d'implantation.
En plus, l'analyse macro et microscopique des rats du groupe 2
(traités par BLM-PDLLA) a montré que les tumeurs
sous-cutanés ont disparus dans 14 rats parmi 21 après traitement.
Le volume de la tumeur a commencé de diminuer 4 jours après
traitement jusqu'à sa disparition complète après 27 jours.
Par contre, dans le groupe 1 (traités par BLM-SOL), une réponse
micro et macroscopique a été détectée dans
seulement 5 rats parmi 21, et le volume de la tumeur augmente
régulièrement après traitement et 21 rats ont
trouvés la mort dans 15 jours comme un résultat de
métastase systémique de la tumeur.
En conclusion, Toru Kumanohoso et al. (T. Kumanohoso,
1997) ont montré que la formulation BLM-PDLLA libère la
bléomycine dans les tissus prés du site d'implantation ainsi que
dans les ganglions lymphatiques régionaux sur une longue période
de temps, et elle a montrée une grande efficacité
thérapeutique que la solution BLM-SOL. Ces résultats indiquent
que le mélange BLM-PDLLA pourrait être une méthode utile
pour un traitement chimiothérapeutique loco- régional.
II.5.2.1.2. Ciblage d'un agent anticancéreux
hydrophobe (Paclitaxel) par l'utilisation d'un copolymère amphiphile
(PVP-b-PDLLA)
Le paclitaxel est une molécule modèle pour le
traitement du cancer. À cause de sa faible solubilité dans l'eau,
le paclitaxel (PTX) a été mélangé avec l'huile de
recin polyoxyélhylée (CrEL) pour la préparation d'une
nouvelle formulation appelée Taxol (c'est la forme commerciale de PTX).
Cette dernière a été utilisée en 1985 pour le
traitement du cancer de l'ovaire réfractaire (D. J. I. Kingston,
1995).
La résistance des cellules anticancéreuses
peuvent limiter l'efficacité du PTX en provoquant le passage rapide de
PTX à travers la membrane cytoplasmique (i.e. diminution de la
concentration de PTX autour de site d'action) (E. K. Rowinsky,
2006). En plus, la formulation Taxol présente un certain nombre
de problèmes, à savoir les patients ont eu une réaction de
choc anaphylactique et d'hypersensibilité sévère
caractérisée par la dyspnée, l'urticaire,
l'angioédème et l'hypotension (H. Gelderblom,
2001).
Afin de résoudre certains défis liés
à la formulation de paclitaxel hydrophobe et pour minimiser des
réactions d'hypersensibilité sévères de surfactif
CrEL formant le Taxol, des nanoparticules à base de copolymère
amphiphile (PVP-b-PDLLA) chargées de PTX ont été
développées.
Genevière Gaucher et al. (G. Gaucher,
2007), ont préparé des nanoparticules de PDLLA
chargées de PTX et entourées d'une couronne hydrophile de PVP.
Ces nanoparticules sont préparées par la méthode
d'émulsion huile/eau (H/E) suivi par évaporation de solvant.
Différentes techniques ont été utilisées pour
caractériser l'interaction entre la matrice polymérique et le
principe actif ainsi que pour déterminer l'efficacité de ces
nanoparticules dans le contrôle de la libération du principe actif
dans le milieu physiologique. Parmi ces méthodes, citant la DRX, la DSC
et le test de dissolution.
L'étude du profil de libération in vitro de PTX
à partir des nanoparticules dans un milieu physiologique (pH = 7,4
à 37 °C) a montré une libération de PTX rapide au
début qui pourrait être attribué à la
présence de molécules du PTX près de la surface des
nanoparticules. Ensuite, la vitesse de libération de PTX devient
inversement proportionnelle à la masse molaire de PDLLA formant le coeur
des nanoparticules. En effet, le PDLLA de masse molaire plus
élevée crée un environnement plus
visqueux menant à une meilleure rétention de PTX dans le vecteur
et à une libération prolongée sur plusieurs jours (figure
51) (G. Gaucher, 2007 ; T. Niwa, 1993).

PDLLA 13400 PDLLA 22000 PDLLA 40500
Temps (jours)
Figure 51. Cinétique de
libération de paclitaxel à partir des nanoparticules de PDLLA
préparées dans le
dichlorométhane comme solvant
organique.
À la lumière des résultats de
libération in vitro du PTX, les nanoparticules à base de PDLLA
ont été sélectionnées pour les expériences
de pharmacocinétique et de biodistribution. Un double marquage à
la radioactivité a été appliqué afin de suivre le
vecteur et le principe actif simultanément dans le sang et dans les
organes. Le PTX solubilisé dans les micelles de CrEL (Taxol) a
été inclus dans l'étude comme formulation
contrôlée. Ces expériences ont été
essayées sur des rats. Les résultats ont montré que les
nanoparticules de PDLLA confères des temps de circulation plasmatique
prolongé au paclitaxel chez l'animal. Ces nanoparticules de PDLLA ont
permis un ciblage du PTX au niveau des tissus tumoraux supérieurs
comparées à la forme commerciale (Taxol).
En résumé, l'incorporation de paclitaxel dans de
nanoparticules de PDLLA est possible et que ces nanoparticules ont
assurées une libération in vitro contrôlée sur
plusieurs jours. Enfin, les nanoparticules polymères de PDLLA
constitueraient une stratégie de vectorisation mieux adaptée au
ciblage tumoral du PTX.
II.5.2.2. Application de poly(D,L-acide lactique) dans le
domaine de l'antibiothérapie
À la différence de l'antibiothérapie
générale, l'antibiothérapie locale est essentiellement
limitée aux sites opératoires, afin de prévenir et/ou de
guérir les infections locales qu'il faut toujours redouter après
une intervention chirurgicale. On connaît les inconvénients de
l'antibiothérapie générale : injections douloureuses avec
irritation des tissus, toxicité rénale ou hépatique et
nécessité d'administrations répétées
(V. Michel, 1993).
Au début, les chirurgiens utilisaient des
biomatériaux implantés sur le site opératoire avant suture
de la plaie chirurgicale, mais l'adhérence bactérienne aux
biomatériaux ainsi que la participation de plusieurs microorganismes
dans la formation de bio-films sur des corps
étrangers ont été à l'origine de
divers complications. Ces dernières se manifestent par des infections et
une intégration insuffisante des implants, ce qui a limité
l'utilisation des biomatériaux dans le corps humain (H.
Gollwitzer, 2003). Même le traitement de ces infections a
généré des problèmes pour le patient ainsi que pour
l'établissement sanitaire : long séjour dans l'hôpital,
augmentation de la morbidité et de la mortalité et des
conséquences économiques très sérieuses (C.
K. Hebert, 1996).
Pour remédier à ces inconvénients
cités précédemment, des compositions biorésorbables
destinées à être implantées sur le site
opératoire, avant suture de la plaie chirurgicale, ont été
utilisées. Elles permettent de mettre en oeuvre une
antibiothérapie interne locale par libération progressive de la
substance antibiotique (V. Michel, 1993).
Hans Gollwitzer et al. (H. Gollwitzer, 2003)
ont proposé d'étudier les propriétés
antibactériennes du poly(D,L-acide lactique) de faible poids
moléculaire contenant le principe actif (gentamycine). Les implants
médicales utilisés sont de nature acier inoxydable et alliage de
Titane (TiAl6V4). La bactérie choisie est l'un des microbes
pathogènes les plus important appelée Staphylococcus
epidermidis (ou S. epidermidis).
La déposition du mélange PDLLA-gentamycine
(dissous dans l'acétate d'éthyle) sur les implants a
été réalisé par la technique dite « solvent
casting ». Plusieurs techniques ont été utilisées
pour caractériser l'effet de cette enveloppe biodégradable et
biocompatible sur la réduction du contact implant-bactérie. Des
tests de dissolution in vitro de l'antibiotique à partir des implants
enveloppés de PDLLA (pH 7,4 à 37 °C), comptage de la
quantité totale de bactérie adhérées à la
surface des implants par radio-étiquetage et la microscopie
électronique à balayage (MEB) pour voir la morphologie de la
surface externe des implants.
i. L'analyse MEB a révélé que l'enveloppe
PDLLA des implants reste stable et que les bactéries sont
complètement détachés des implants.
ii. Le test de libération de gentamycine à
partir des implants dans un milieu physiologique (pH = 7,4 à 37 °C)
a montré que la libération de gentamycine se fait d'une
manière continue pour au moins 96 heures. La figure 52 montre le profil
de libération de gentamycine à partir de chaque
échantillon. Le graphe montre une libération rapide de
l'antibiotique dans les premières heures suivie d'une libération
lente et continue dans le reste du temps. La concentration de gentamycine de
l'échantillon non traité reste constante sur tout l'intervalle de
temps de l'étude.
iii. Le nombre total de bactéries adhérentes a
augmenté dans tout les échantillons
enveloppés par le
PDLLA, mais le nombre de bactéries capables de se
développés a diminué dans les implants enveloppés
par le PDLLA comparativement aux implants non traités.

Temps (h)
Figure 52. Profils de libération de
gentamycine à partir de l'acier inoxydable recouvert de PDLLA.
En résumé, l'enrobage des implants
médicaux par le PDLLA a augmenté le nombre total de
microorganismes attachés aux implants, mais au même temps il a
réduit significativement le nombre de bactéries vivantes
même sans l'antibiotique, suggérant que l'enveloppe de PDLLA pur
présente une action bactéricide contre S. epidermidis.
Enfin, l'incorporation de gentamycine dans le PDLLA donne un système de
vectorisation de médicaments local.
II.5.2.3. Application dermato-thérapeutique de
poly(D,L-acide lactique)
La dermatologie est une partie de la médecine qui
étudie et soigne les maladies de la peau et ses anomalies
inesthétiques. Les formes galéniques classiques appliquées
sur la peau peuvent pénétrer à travers la stratum corneum
et les follicules pileux sans distinction. Des maladies associées aux
follicules pileux peuvent apparaitre, comme acné vulgarise qui est due
principalement à un disfonctionnement des glandes sébacées
situées à la base des follicules pileux de la peau.
Des micro et nanoparticules ont été
étudiées pour le traitement dermatologique depuis des
décennies et certains de ces formulations sont déjà
commercialisés. Plusieurs types de particules sont actuellement
utilisées comme vecteurs de principes actifs pour des applications
topiques, à savoir liposomes modifies, nanoparticules lipidique solides
et des polyesters biodégradables comme PLA, PLGA et PCL. Les
nanoparticules sont très intéressantes pour des applications
dermatologiques à cause de leur tendances a
pénétrés et à s'accumulers dans les orifices des
follicules pileux de la peau. C'est pourquoi les nanoparticules ont
été proposées pour la délivrance spécifique
de médicaments vers les structures pilosébacés (F.
Rancan, 2009). L'application des particules chargées de
principes actifs dans les traitements dermatologiques implique la
réduction des voies transépidermales et l'augmentation de la
concentration en principes actifs dans les follicules pileux (J.
Lademann, 2008). Cette accumulation préférentielle
implique une amélioration de l'effet thérapeutique des principes
actifs dans la dermatologie, en
particulier, dans la thérapie des maladies
associées aux follicules pileux de la peau (F. Rancan,
2009). En effet, le dépôt de particules dans les
follicules pileux assurent une libération prolongée de principes
actifs, ce qui permet la réduction de la dose et d'éviter
l'administration répétée de médicaments (M.
Schäfer-Korting, 2007).
Rancan et al. (F. Rancan, 2009), ont
étudié la convenance des nanoparticules à base de
poly(D,L-acide lactique) comme un vecteur pour la vectorisation
transépidermale de principes actifs. Pour se faire, deux types de
nanoparticules de PDLLA ont été utilisées : des particules
de 228 nm de diamètre chargées avec rouge de nile (NR_PDLLA_228)
et des particules de 365 nm de diamètre chargées avec la
coumarine-6 (Coum_PDLA_365). Rouge de nile est choisi à cause de son
émission dans le spectre du visible où la peau présente
une faible auto-fluorescence et la coumarine-6 est choisi à cause de sa
haute fluorescence.
Le profile de pénétration des nanoparticules de
PDLLA dans les follicules pileux de la peau chez l'homme, la libération
du colorant incorporé et la perméation du colorant dans
l'épiderme et la structure pilosébacé ont
été étudiés.
Les nanoparticules de PDLLA (30 000 g/mol) ont
été préparées par la méthode
d'émulsion huile/eau suivie de l'évaporation du solvant. La peau
humaine a été obtenue, après 24 heures après
excision chirurgicale, à partir des volontaires en bonne santé
subissant une chirurgie plastique. Des colorants fluorescents ont
été incorporés dans les particules de PDLLA dans le but de
faciliter la détection microscopique des particules, comme les
composés lipophiles modèles pour l'étude de la
libération des substances incorporés d'une part, et leur
diffusion hors des particules ainsi que leur rétention dans les
différents compartiments de la peau, d'autre part.
Rancan et al. (F. Rancan, 2009), ont
trouvé que des particules de PDLLA se sont accumulées
préférentiellement dans les follicules pileux, tel que,
approximativement 50 % des follicules pileux analysées sont
occupées par les particules de PDLLA. Dans 12-15 % des follicules
pileux, les particules de PDLLA ont pénétrées dans
l'infundibulum et ont été également observées dans
l'entrée des glandes sébacés. L'accumulation des
particules dans les follicules pileux a été accompagnée
par la libération des colorants dans l'épiderme viable et leur
rétention dans les glandes sébacés pour 24 heures. Le test
de libération in vitro des colorants dans un système biphasique,
consistant à un solvant organique lipophile et une solution tampon, a
été effectué. La cinétique de libération de
rouge de nile et coumarine-6 à partir des particules de PDLLA a
montrée une libération rapide dans la phase organique suivi par
un plateau après environ 16 heures (figure 53).
La libération rapide de NR et Coum-6 à partir
des nanoparticules de PDLLA après contact avec le solvant lipophile peut
être due à la partition des colorants entre le noyau lipophile des
particules et le solvant lipophile ainsi que la dissolution de polymère
dans la phase lipophile. Par contre la libération lente des colorants
à partir des particules dans la phase aqueuse est due à
l'hydrolyse de polymère et à l'érosion des particules. La
concentration locale élevée atteinte par les nanoparticules
accumulées dans la tranche de la peau et les conduites folliculaires
pourrait donc favoriser l'agrégation des particules
dénaturées.

Temps (heure)
Figure 53. Libération in vitro de nile
rouge (NR) et coumarine-6 (Coum-6) à partir des particules de
PDLLA
dans un système biphasique (hexane/solution tampon).
En conclusion, la formation des agrégats de particules,
qui sont observés dans le test in vitro et in vivo, suggère que
les particules de PDLLA, à la différence des particules solides
stables comme les nanosphères de polystyrène, ne s'accumulent pas
seulement, mais aussi se déstabilisent et libèrent son contenu en
colorant tout et en formant des agrégats dans les conduites des
follicules pileux. Par conséquent, les particules de PDLLA peuvent
être des candidats idéals pour la conception des systèmes
de vectorisation de médicaments dans le but d'assurer le ciblage de
principes actifs dans les follicules pileux (F. Rancan,
2009).
En résumé
Ce deuxième chapitre bibliographique nous a permis de
décrire le concept général de la vectorisation de
principes actifs au moyen des polymères et en particulier, ceux à
caractère biodégradable. Nous avons décrit les
différents systèmes ainsi que les différentes
architectures et les types de polymères exploités dans le domaine
de la vectorisation de médicaments. On a pu constater que dans la
plupart des cas, il s'agit de copolymères à bloc et que les
architectures griffées sont assez peu nombreuses. Enfin, nous avons
clôturé ce chapitre par quelques exemples d'applications
récentes de poly(D,L-acide lactique) (PDLLA) dans le domaine
pharmaceutique, en particulier dans le domaine de vectorisation de
médicaments. On a pu constater que le PDLLA est très
utilisé comme vecteurs de médicaments grâce à ces
propriétés de biodégradabilité,
biorésorbabilité et biodisponibilité. On a
constaté, également, que le poly(D,L-acide lactique) peut
être utilisé comme homopolymère ou sous forme d'un bloc
copolymère amphiphile (ex. PLGA, PDLLA-PVP ...).
SECTION EXPÉRIMENTALE
Chapitre III. Matériaux et méthodes
utilisées
Chapitre III. Matériaux et méthodes
utilisées
Partie I. Synthèse de poly(D,L-acide
lactique) par la méthode de polycondensation
azéotropique
Introduction
L'existence des groupements fonctionnels -OH et -COOH dans la
molécule d'acide lactique lui permet d'être converti directement
en un polyester par polycondensation. Cependant, cette réaction ne
permet pas d'avoir des masses moléculaires assez élevées,
à cause des difficultés inhérentes conduisant
l'équilibre de déshydratation dans la direction de
l'estérification. Pour surmonter cette difficulté, l'utilisation
de solvants organiques pour la distillation azéotropique de l'eau de
condensation, en présence d'un catalyseurs, permet le contrôle de
l'équilibre entre acide lactique, H2O et PLA, et par conséquent
des masses moléculaires relativement élevées peuvent
être obtenues (K. M. Nampothiri, 2010).
Ajioka et al. (M. Ajioka, 1998), ont
montré que des masses moléculaires élevées de PLA
peuvent être obtenues par une étape de polycondensation, à
condition que des solvants organiques azéotropiques sont
employés.
Pour une application de PLA dans le domaine pharmaceutique,
des caractéristiques de biodégradabilité et de de
biorésorbabilité sont exigées, par contre la
résistance mécanique n'a pas d'importance particulière.
Dans ce cas, des masses moléculaires de l'ordre de 2 à
3.103 Da semblent plus adéquates pour de telles applications
(C. S. Proikakis, 2002).
I.1. Matériaux utilisés
Le D,L-acide lactique (85 %), ayant pour formule brute
C3H6O3 et une masse molaire de 90,08 g/mol, produit par la compagnie
BIOCHEM Chemopharma, a été utilisé. C'est un liquide de
couleur jaune et corrosif, possède un point de fusion de 16,8 °C et
un point d'ébullition de 122 °C (à 12 mmHg).
Le Xylène, ayant pour formule brute C8H10
et une masse molaire de 106,16 g/mol, a été utilisé comme
solvant azéotropique. C'est un liquide incolore avec une densité
de 0,861 g/cm3, possédant un point de fusion de 13,2 °C
et un point d'ébullition de 138,35 °C. Il est très soluble
dans l'éthanol et dans le diéthyle éther et insoluble dans
l'eau.
Le Chlorure d'étain dihydraté (SnCl2,2H2O), de
masse molaire de 225,63 g/mol et d'une densité de 2,71 g/cm3
a été utilisé comme catalyseur. C'est un solide cristallin
de couleur blanche et corrosif produit par la compagnie BIOCHEM Chemopharma. Il
possède un point de fusion de 37 °C et un point d'ébullition
de 623 °C. Il est soluble dans plusieurs solvants organiques
(éthanol, acétone et l'éther) et insoluble dans le
xylène.
I.2. Protocole expérimental
100 ml d'une solution de D,L-acide lactique a
été versée dans un ballon tri cols de 500 ml. Dans le but
d'éliminer les molécules d'eau présentes dans la solution,
cette dernière a été chauffée jusqu'à 100
°C pendant 24 h. Ensuite, un volume de 250 ml de xylène et une
quantité de chlorure d'étain dihydraté équivalente
à 0,2 % de la masse de la solution D,L- acide lactique ont
été ajoutées au mélange résiduel.
Après élimination d'oxygène présent dans la
solution par barbotage avec un gaz inerte (Argan), la réaction de
polycondensation s'est effectuée à une température de 138
°C pendant un temps déterminé. La température de la
réaction a été ajustée à l'aide d'un
régulateur de température muni d'un thermocouple.
L'élimination de l'eau produite durant la polycondensation s'effectue,
durant toute la durée de la réaction, par distillation
azéotropique avec le xylène (figure 54) (C. S. Proikakis,
2002 ; K. W. Kim, 2002).
Pour synthétiser des polymères PDLLA de
différentes masses moléculaires, on a choisitrois
temps de réaction à savoir 41, 55 et 65 heures. Une fois la
polycondensation terminée, un
mélange visqueux a été obtenu
après refroidissement à l'air ambiant. L'addition continue du
méthanol (solvant de précipitation pour le PLA) dans le
mélange visqueux a permis de faire précipiter le polymère.
Le produit blanc obtenu a été récupéré par
filtration et laissé sécher à l'étuve à 40
°C. Enfin, comme les produits de synthèse son
généralement non pur, une procédure de recristallisation
du polymère obtenu a été nécessaire. Le
polymère a été dissous dans du chloroforme, ensuite une
addition continue du méthanol a permet d'avoir un
précipité blanc et après séchage le produit sec
obtenu a été réduit en poudre.

N2
Thermomètre
Gaz (N2)
Réfrigérant
N2
Plaque chauffante
Figure 54. Montage de polymérisation
azéotropique de D,L-acide lactique dans une atmosphère inerte.
I.4. Mesure de la masse viscosimétrique des
polymères obtenus
Les viscosités de solutions de PDLLA de
différentes concentrations préparées, allant de 0,2
à 0,5 g/dl ont été mesurées à l'aide d'un
viscosimètre capillaire de type Ubbelhode dans un bain d'eau
thermostaté à 30 °C. Le chloroforme a été
utilisé comme solvant de dissolution du PDLLA. Les temps
d'écoulement pour le chloroforme (t0) et pour les solutions de
différentes concentrations (t) ont été mesurées.
Les viscosités relatives (ir) ont été alors
obtenues à partir
des rapports de t0/t, et les viscosités
spécifiques (rsp), qui est égale à ln
rr /c, ont été tracées en fonction des
concentrations préparées (c). L'intersection de la droite rsp =
f(c) obtenue avec l'axe des ordonnées après extrapolation, nous a
donné la valeur de la viscosité intrinsèque, [r]. La masse
viscosimétrique de chaque polymère est donnée par
l'équation de Mark - Houwink donnée (1) ci-dessous (A. G.
Andreopoulos, 1999 ; X. Kaitian, 1996).
[ç] = KMa (1)
où M est la masse moléculaire du
polymère, K et a sont des constantes pour un systèmes
polymère-solvant à une température donné. Dans le
cas du système PDLLA-Chloroforme à 30 °C, les valeurs de a
et K sont 0,82 et 1,29.10-4 ml/g, respectivement (K. W. Kim,
2002).
Partie II. Préparation des formulations
polymère/principe actif
Introduction
La voie orale est la voie d'administration de choix des
médicaments car elle est la plus naturelle et la plus confortable pour
le patient, permettant de garantir au mieux le suivi des prescriptions. Les
formulations solides orales possèdent plusieurs avantages par rapport
aux autres formes orales.
La libération du principe actif est une étape
cruciale et déterminante pour la biodisponibilité orale du
principe actif, particulièrement pour ceux de faible solubilité
et de haute perméabilité gastro-intestinale. Par
l'amélioration du profile de libération de ces principes actifs,
il est possible d'augmenter leur biodisponibilité et de réduire
les effets secondaires (T. Vasconcelos, 2007).
De nombreuses formulations à base de polymères
ont été proposées. Cependant, les formulations
préparées par la technique de dispersion solide ont
été les plus utilisées. La structure des particules
polymères chargées en principe actif dépend du
procédé de fabrication utilisé, de la nature des
matériaux polymères et du caractère lipo- ou hydrosoluble
de principe actif (A. Wawrezinieck, 2008). Dans notre travail,
les dispersions solides ont été préparées à
l'aide de plusieurs méthodes, à savoir mélange physique,
mélange par fusion et mélange par évaporation de
solvant.
II.1. Matériaux utilisés
Les produits utilisés dans notre préparation
sont le Poly(D,L-acide lactique) synthétisé dans le laboratoire
des matériaux organiques (LMO) de l'Université de Bejaïa.
L'ibuprofène offert gracieusement par le groupe SAIDAL que nous
remerciant. Le dihydrogénophosphate de potassium, ayant pour formule
générale KH2PO4 et une masse molaire de 136,09 g/mol produit par
PANREAC QUIMICA SA ainsi que l'hydroxyde de sodium (NaOH), ayant une masse
molaire de 40,00 g/mol, produit par le groupe Merck KGaA (Germany) sont
utilisés pour ajuster le pH des milieux physiologiques.
II.2. Protocoles expérimentales
Pour toutes les préparations, une quantité de
1,5 g d'ibuprofène (IB) a été mélangée avec
le polymère PDLLA à différents pourcentages. Le tableau 14
suivant résume le contenu des formulations préparées.
Tableau 14. Constitution des formulations
préparées.
|
Formulation
|
F5
|
F15
|
F25
|
|
Ibuprofène (g)
|
1,5
|
1,5
|
1,5
|
|
Poly(D,L-acide lactique) (%)
|
5
|
15
|
25
|
II.3.1. Préparation des mélanges physiques
IB/PDLLA
Nous avons mélangé l'ibuprofène avec le
poly(D,L-acide lactique) dans un mortier pendant environ 15 min (B.
Luppi, 2008).
II.3.2. Préparation des mélanges IB/PDLLA par
évaporation de solvant
La méthode d'évaporation de solvant consiste
à solubiliser le principe actif et le polymère dans un solvant
volatile approprié (commun) suivi par une évaporation de ce
dernier. Dans un erlenmeyer, nous avons fait dissoudre 1,5 g
d'ibuprofène avec une quantité de PDLLA (5, 15 et 25 %) de la
masse d'IB dans 150 ml de diméthylformamide (DMF). Le mélange est
laissé sous agitation magnétique pendant 30 heures. Ensuite, une
procédure d'extraction de solvant sous vide, à une
température de 65 °C, a été effectuée (figure
55). Un résidu de couleur blanche au fond de l'erlenmeyer a
été obtenu, il a été réduit en poudre
après séchage (A. C.
Williams, 2005).

Bain-marie
65 °C
Vide
Bain d'eau glacée
Mélange
IB + PDLLA + DMF
Figure 55. Montage de la méthode
d'évaporation de solvant sous vide.
II.3.3. Préparation des mélanges IB/PDLLA par
fusion à chaud
La méthode de fusion consiste à faire fondre le
mélange principe actif/polymère suivi par un refroidissement et
une pulvérisation du produit obtenu.
Dans un erlenmeyer, un mélange de 1,5 g
d'ibuprofène avec le poly(D,L-acide lactique) différents
pourcentages (5, 15 et 25 %) a été préparé.
Ensuite, nous avons chauffé le mélange, à l'aide d'une
plaque chauffante menée d'un régulateur de température
jusqu'à 120 °C sous une agitation magnétique pendant 30
minutes (figure 56). Un mélange pâteux de couleur blanche a
été obtenu. Enfin, nous avons laissé le mélange se
refroidir à température ambiante pendant 24 heures puis on l'a
réduit en poudre (A. C. Williams, 2005).
|
|
Mélange IB + PDLLA
Bain d'huile (120 °C) (Glycérol)
Plaque chauffante
|
|
Figure 56. Montage de la méthode de
fusion à chaud.
II.4. Techniques de caractérisation des
formulations
II.4.1. Analyse thermique
II.4.1.1. Analyse thermique (ATG - ATD)
L'analyse thermogravimétrique (ATG) consiste à
mesurer la variation de masse d'un échantillon lors d'un cycle
thermique. L'analyse thermique différentielle (ATD) est basée sur
l'étude de l'énergie dégagée ou absorbée par
le matériau quand il subit des transformations physiques ou chimiques
lors d'un cycle thermique. Ces analyses sont relatives. Les valeurs obtenues
sont issues de l'évolution des paramètres de l'échantillon
comparativement à celle d'une référence étalon
inerte.
Dans ce travail, les échantillons de masses 11 à
22 mg ont été placés dans un four d'un analyseur
thermo-gravimétrique (SETARAM TG - DT A92) sous une atmosphère
inerte (N2) pour prévenir l'oxydation. La température a
été variée de 20 à 500 °C avec un
incrément de 10 °C/min.
II.4.1.2. Analyse thermique différentielle
L'analyse thermique différentielle permet de
déterminer les températures de transition vitreuses
(Tg), de cristallisation (Tc) et de fusion (Tf) pour
chaque polymère synthétisé. Le principe de la mesure
repose sur la différence de chaleur que l'on doit fournir à un
échantillon donné par rapport à une
référence inerte pour les amener à une même
température.
Un appareil de DSC (TA Instruments DSC 2920) a
été utilisé. Les échantillons de masses 8,5
à 9,5 mg ont été placés dans des creusets en
aluminium et ont été analysés sur une plage de
température de 20 à 180 °C pour le PDLLA et de 25 à
110 °C pour l'ibuprofène, sous atmosphère d'azote, avec une
vitesse de montée en température de 10 °C/min.
II.4.2. Analyse spectrale
II.4.1.1. Spectrométrie infrarouge à
transformée de Fourier (IRTF)
Nous avons utilisé un spectrophotomètre
infrarouge à transformé de Fourier IRAffinity-1 CE Shimadzu,
(Japan). L'analyse par spectroscopie IRTF a été effectuée
sur des pastilles obtenues en ajoutant 1,5 mg du mélange IB/PDLLA dans
100 mg de KBr. Le spectre IRTF de chaque échantillon est
enregistré à la température ambiante dans la plage de
400-4000 cm-1.
II.4.2.2. Diffraction des rayons X (DRX)
Les analyses de diffraction des rayons X ont été
réalisées avec un diffractomètre expert prof panalytical.
La longueur d'onde de la radiation utilisée est celle du
Ká1= 1,5406 Å. Elle est générée
par une anode en cuivre, sous une tension de 45 kV et un courant de 30 mA et un
monochromateur constitué par un monocristal de NO. Les
échantillons sont préparés par pressage manuel dans des
petits cylindres plats. L'acquisition du diffractogramme est effectuée
à des angles 2è compris entre 5 et 50 °. Le type de
balayage est continu avec un pas de 0,02 ° et une vitesse de 7
°/min.
II.4.2.3. Microscopie électronique à
balayage (MEB)
La microscopie électronique à balayage est
employée pour déterminer les changements de morphologie de taille
et d'agglomération des échantillons. La morphologie de surface et
de section des granules enrobés ou non a été
observée par MEB (FEG 600) en mode pression variable, muni d'un
détecteur LFD.
II.4.2.4. Spectrophotométrie UV-Visible
Le principe de la spectrophotométrie UV-Visible repose
sur la transition d'un état fondamentale vers un état
excité d'un électron d'une molécule excité par une
onde électro - magnétique. Le passage d'un état
électronique à un autre état électronique
d'énergie plus élevée nécessite l'absorption
d'énergie sous forme de photons. Un spectromètre consiste en une
source constituée de deux lampes qui permettent un continuum
d'émission sur toute la gamme de longueur d'onde UV-Visible :
- Lampe au deutérium qui émet des longueurs d'ondes
de 180 à 400 nm (UV).
- Lampe au tungstène qui émet des longueurs d'ondes
de 400 à 800 nm (Visible).
Dans notre travail, nous avons utilisé un
spectrophotomètre UV-Visible (Optizen 2120UV) pour le dosage du principe
actif durant les tests de dissolution en utilisant la fameuse loi de
Beer-Lambert-Bouguer (2) suivante :
A = å × l ×C (2)
Où A = Absorbance
å = Coefficient d'absorption
(l.mol-1.cm-1) l = Longueur de la cuve (cm)
C = Concentration de l'échantillon
(mol.l-1).
II.5. Etude de dissolution in vitro des
échantillons
II.5.1. Préparation du milieu tampon
Dans un récipient en verre, une masse de 2 g de KH2PO4
a été ajoutée à 3 litre d'eau distillée.
Ensuite, nous avons mesuré le pH de la solution obtenue à l'aide
d'un pH-mètre (HANNA pH 211) et une valeur de pH = 4,7 est obtenue.
Enfin, à l'aide d'une burette, nous avons ajusté la valeur du pH
par l'ajout d'une solution de NaOH (2 g/l) jusqu'à avoir le pH
souhaité (5,8 et 7,4).
II.5.2. Courbes d'étalonnage de l'ibuprofène
à chaque valeur de pH
Dans un bécher de 1 litre, un mélange de 20 mg
d'ibuprofène avec 900 mL du milieu tampon (pH 7,4 ou 5,8) a
été préparé. Ensuite, à partir de cette
solution mère, plusieurs solutions de concentrations différentes
en TB ont été préparées par dilution. Enfin, nous
avons déterminé les valeurs des absorbances (A) correspondantes
aux concentrations (C) préparées à
l'aide d'un spectrophotomètre UV-Visible. Le tracé
de A = f (C) nous a permet d'avoir les courbes d'étalonnage
correspondantes (figures 57 et 58).

1
A = 0,0403 * C
R2 = 0,9988
0,8
0,6
0,4
0,2
0
Absorbance
0 5 10 15 20
Concentration (mg/900 ml)
Figure 57. Courbe d'étalonnage de
l'ibuprofène à pH 7,4.
0 5 10 15 20
Concentration (mg/900 ml)

Absorbance
1
0,8
A = 0,0341 * C
R2 = 0,9964
0,6
0,4
0,2
0
Figure 58. Courbe d'étalonnage de
l'ibuprofène à pH 5,8.
II.5.3. Préparation des comprimés
La fabrication des comprimés à base des
échantillons préparés s'est réalisée
à l'aide d'une comprimeuse manuelle. Pour l'ibuprofène pur, une
masse de 20 mg a été pesée à l'aide d'une balance
analytique. La poudre pesée a été ensuite versée
dans un moule, à l'aide d'une presse hydraulique manuelle on a pu
transformer la poudre en un comprimé solide non friable. La même
procédure a été utilisée pour la préparation
des comprimés à base d'échantillons enrobés tout en
respectant la constitution de chaque formulation. Le tableau 15 suivant
résume la constitution de chaque comprimé.
Tableau 15. Constitution des comprimés
|
Formulation
|
F5
|
F15
|
F25
|
|
IB (mg)
|
20
|
20
|
20
|
|
PDLLA (%)
|
5
|
15
|
25
|
|
PDLLA (mg)
|
1
|
3
|
5
|
II.4.4.4. Test de dissolution
Les essais de dissolution du principe actif ont
été réalisés à l'aide d'un appareil à
pales tournantes (Pharma Test DT70, Germany) (figure 59). Les
échantillons (sous formes de comprimés) sont soumis à une
agitation constante dans 900 mL de milieu de dissolution préparé
précédemment, à une température constante de 37
°C (#177; 0,5 °C). La vitesse de rotation des pales est fixée
à 50 tr/min. Aux temps prédéterminés, des
échantillons de 3 ml du milieu ont été
prélevés et une quantité équivalente du milieu
tampon fraîche a été ajoutée de nouveau au milieu de
dissolution pour maintenir le volume constant. Le contenu en ibuprofène
de chaque échantillon a été dosé par
spectrophotométrie UV-Visible à 223 nm.
Système de prélèvement
|
Milieu de
dissolution
|
|
|
|
Pale
|
|
|
|
Bol
|
|
|
Comprimé
|
|
|
|
|
Figure 59. Représentation
schématique d'une cellule du dissolutest.
Chapitre IV. Résultats et discussions
Chapitre IV. Résultats et discussions
Partie I. Synthèse et caractérisation de poly(D,L-acide
lactique)
I.1. Synthèse de PDLLA
Dans ce travail, des homopolymères de type
poly(D,L-acide lactique) avec des masses moléculaires faibles ont
été synthétisés par la méthode de
polymérisation par polycondensation azéotropique du
monomère D,L-acide lactique selon le schéma 5 global suivant.

i)
Prédistillation
à 100 °C (24 h)
iii) Polymérisation en présence SnCl2,2H2O et
p-xylène à 140 °C
OH
n
HO
O
n
*
*
O
O
Schéma 5. Réaction de
polymérisation par polycondensation azéotropique.
Les valeurs des masses moléculaires
viscosimétriques moyennes (Mv) des PDLLA
synthétisés à différents temps de
polymérisation sont regroupées dans le tableau 16 suivant.
Tableau 16. Les masses viscosimétriques
de PLA pour chaque temps de polymérisation.
|
Temps (h)
|
Masse viscosimétrique (Mv) de PDLLA
(Da)
|
|
41
|
9000
|
|
55
|
3000
|
|
65
|
1000
|
Les rendements des homopolymères, qui est défini
comme le rapport « quantité de monomère/quantité de
polymère », obtenus par la polycondensation directe de D,L-acide
lactique étaient autour de 40-50 %.
Pour la synthèse de poly(D,L-acide lactique), nous
avons utilisé une procédure de polycondensation
azéotropique en présence d'un catalyseur de type chlorure
d'étain dihydraté. Cependant, l'effet de SnCl2,2H2O sur le
mécanisme réactionnel de polycondensation n'est pas encore clair.
Yamaoka et al. (T. Yamaoka, 1999) ont proposé un
mécanisme possible de l'activité catalytique des composés
à base d'étain, le mécanisme est constitué de trois
étapes. En effet, la première étape consiste à la
formation de l'oxyde d'étain-(II) (1) par hydrolyse de SnCl2,2H2O.
Ensuite, dans la deuxième étape, le groupement -COOH terminal
d'un oligomère de PLA réagir avec le groupement -OH de (1) pour
former de l'eau et un autre composé à base d'étain (2).
Enfin, dans la troisième étape, le groupement -OH de
l'oligomère devrait être lié par une liaison de
coordination avec le composé (2) pour former un autre composé
à base d'étain (3), de telle sorte qu'une condensation peut
être induit autour de l'atome Sn afin de former le polymère (4) et
une forme active hydratée (schéma 6).
Première étape

O
Cl
O
Cl
Sn
H
+
H
Sn
H
O
H
Cl
Cl
O
H
H
H
H
OH
OH
L
L
OH
HO
Sn
Sn

O
H
H
H
O
Sn
O
H
HCl
H
O
Sn
Sn
O
H
O
H
H
+
Cl
Cl
H
O
+ 2L
Sn
O
H
HCl
+

L
OH
OH
L
HO
C
O
L
OH
OH
C
L
HO
OH
O
Sn
Sn
H
OH
OH Sn
L
L
OH
OH
Sn
O
HO
O
C
H2O
O
Sn
Sn
Deuxième étape
(2)
Troisième étape

OH
L
HO
O Sn
L
OH
HO
OH
OH
L
L
O
Sn
Sn
Sn
O
C
HO
O
C
HO
(3)
OH
L
Sn
Sn
L
HO
H2O
O H O C O
O
(4)
C O
L
OH
L
OH
HO
+
Sn
Sn
Régénération de le forme oxydée du
catalyseur

L
OH
L
HO
OH
OH
Sn
Sn

OH
Sn
Sn
HO
L
L
OH
OH
Schéma 6. Mécanisme
réactionnel de la synthèse de PLA en présence d'un
catalyseur (SnCl2).
I.2. Caractérisation des polymères
obtenus
I.2.1. Analyse thermique
I.2.1.1. Etude TG/DTG
La figure 60 montre les courbes TG/DTG obtenues à 0,5
°C/min sous atmosphère d'azote pour les trois polymères
PDLLA (1000,3000 et 9000).
On remarque que les températures de dégradation
des différents polymères sont compris entre 260 et 270 °C.
Les températures de début et fin de dégradation des trois
polymères sont 226,8-279,4 pour PDLLA1000, 235,3-286,4 °C pour
PDLLA3000 et 228,9-278,3 °C pour PDLLA9000. On remarque que l'intervalle
des températures de début et fin de dégradation pour les
trois polymères est approximativement [230-280] °C. La courbe DTG
obtenu consiste à un seul pic, ce ci indique qu'il ya principalement une
seule étape réactionnelle
durant la dégradation thermique des trois
polymères. En effet, cette constation est probablement associée
avec la perte des groupements ester, ce résultat est en accord avec ce
qui a été déjà fait (A. Nalbandi,
2001).
PDLLA 1000
Td = 252°C
DTG
TG

Td = 268°C
DTG
TG
PDLLA 3000
|
120 100 80
|
|
|
5
0
-5
|
|
60 40 20 0
|
m(%)
|
dm/dt (%/min)
|
-10 -15 -20 -25 -30
|
|
120 100 80 60 40 20 0
|
m (%)
|

|
dm/dt (%/min)
|
5 0 -5 -10 -15 -20 -25 -30
|
0 100 200 300 400 500
Température (°C)
0 100 200 300 400 500
Température (°C)

PDLLA 9000
Td = 254°C
TG
DTG
|
120 100 80 60 40 20 0
|
m (%)
|
|
dm/dt (%/min)
|
0 -5 -10 -15 -20 -25 -30
|
0 100 200 300 400 500
Température (°C)
Figure 60. Courbes de TG/DTG obtenus à
10 °C/min sous atmosphère d'azote pour le poly(D,L -
acide
lactique) de différentes masses moléculaires.
I.2.1.2. Etude DSC
Les thermogrammes représentant les trois
polymères PDLLA de différntes masses sont montrés dans la
figure 61. Les thermogrammes ont été enregistrés par un
calorimètre différentiel à balayage, à une vitesse
de chauffage de 10 °C/min, et dans un intervalle de température de
40 à 180 °C.
Trois transitions thermiques sont observées : (1) le
pic de transition vitreuse Tg, qui est le principal changement
d'état intervenant dans les polymères amorphes, il correspond au
passage d'un état liquide surfondu à un état vitreux lors
du refroidissement dans la région de température de transistion
vitreuse ; (2) le pic de recristallisation (ou cristallisation froide)
éxothermique Tc, qui est dû à une
cristallisation limitée après refroidissement rapide du
polymère fondu, entre Tg et Tc, le PDLLA est dans
un état liquide de viscosité élevée et faiblement
cristallin ; (3) le pic de fusion endothermique Tf, il donne des informations
sur la morphologie. Le pic de fusion donne aussi l'information sur le type de
cristallisation présent dans le polymère. En effet, l'observation
d'un double pic de fusion sur les thermogrammes de PDLLA (1000 et 3000) pourait
indiquer la présence de populations de cristaux de morphologies
différentes ou cristaux avec des imperfections (C. M. Boutry,
2010).
Chapitre IV. Résultats et discussion

20 40 60 80 100 120 140 160 180
40 60 80 100 120 140 160 180
Température (°C) PDLLA 9000
Tf = 140,40
Température (°C)
Endothermique Exothermique
T = 88,77
c
-1,2
20 40 60 80 100 120 140 160 180
PDLLA 3000
Endothermique Exothermique
T = 98,81
c
Tf = 144,66
0,3
0,2
0,1
Flux de chaleur (mW.mg')
0,0
-0,1
-0,2
-0,3
-0,4
-0,5
-0,6
0,3
0,2
0,1
Flux de chaleur (mW.mg')
0,0
-0,1
-0,2
-0,3
-0,4
-0,5
0,2
Flux de chaleur (mW.mg')
0,0
-0,2
-0,4
-0,6
-0,8
-1,0
PDLLA 1000
Endothermique Exothermique
T = 102,76
c
Tf = 140,80
Température (°C)
Figure 61. Les thermogrammes DSC des trois
polymères PDLLA obtenus à une vitesse de chauffage de
10
°C/min.
La méthode DSC est particulièrement utile pour la
détermination du taux de cristallinité des polymères. Le
calcul du taux de cristallinité ,Xc, est donné par la
formule suivante (C. Chen,
2004).
Xc (%) = [AHf
/AHfo] x 100% (3)
avec ÄHf est l'enthalpie de fusion de PLA
déterminé à partir de la surface du pic endothermique.
ÄHfo est l'enthalpie de fusion pour un cristal du
même polymère. Pour le
PLA, ÄHfo est supposé égale
à 93 J/g (C. Chen, 2004 ; M. F. Gonzalez, 1999). Les
résultats données par la technique DSC sont regroupés dans
le tableau 17 suivant.
Tableau 17. Les différentes informations
sur la PDLLA données par DSC.
|
PDLLA (Da)
|
Tg (°C)
|
Tc (°C)
|
Tf (°C)
|
AHf (J/g)
|
AHc (J/g)
|
Xc (%)
|
|
1000
|
/
|
102,76
|
140,80
|
25,11
|
19,92
|
27
|
|
3000
|
/
|
98,81
|
144,81
|
31,35
|
17,80
|
34
|
|
9000
|
/
|
88,77
|
140,40
|
43,77
|
5,84
|
47
|
I.2.2. Analyse structural
I.2.2.1. Spectres IRTF des polymères obtenus
Les spectres IR-TF des trois polymères PDLLA sont
montrés dans la figure 62. Les trois spectres montrent une bande large
à 1759 cm-1 qui est due à la vibration
d'élongation de la liaison du groupe carbonyle ester C=O, des bandes
situées à 3001 cm-1 et 2953 cm-1 qui sont
dues à la vibration d'élongation de la liaison C-H du groupe CH3.
D'autres bandes sont observées dans la région 1300-1050
cm-1 qui sont caractéristiques de la vibration
d'élongation de la liaison C-O du groupe ester (la bande la plus
caractéristique dans ce cas est celle qui apparait à 1188
cm-1). Le pic à 3508 cm-1 est due à la
vibration d'élongation de l'hydroxyle (O-H) terminal. L'apparition de ce
pic peut être utilisé comme une indication des polymères de
faibles masses moléculaires (Z. Zhong-Cheng, 2005 ; X. Kaitian,
1996).

Transmittance (%)
(a)
(c)
(b)
OH
3508
CH3
3001
2953
C=O
1759
1456
1362
C-O
11881134
1134
871
757
695
4000 3500 3000 2500 2000 1500 1000 500
Longueur d'onde (cm-1)
Figure 62. Les spectres IRTF des trois
polymères PDLLA : (a) PDLLA 1000, (b) PDLLA 3000 et (c) PDLLA
9000
Da).
I.2.2.2. Spectres DRX des PDLLA obtenus
Le degré de cristallinité est l'un des
paramètres de base les plus important dans la caractérisation des
polymères semicristallins. En effet, la cristallinité affecte
presque toutes les properiétés des polymères, à
savoir les properiétés mécaniques, physiques,
thermodynamiques et optiques (J. Bidone, 2009).
Les spectres DRX des trois polymères PDLLA sont
donnés dans la figure 63. Les pics de diffraction
caractéristiques de poly(D,L-acide lactique) sont ceux prévus
pour la forme á de PDLLA (maille orthorhombique de paramètres a =
10,6, b = 6,1 et c = 28,8 Å) (J. Cho, 2003 ; W. Hoogsteen,
1990). Les spectres DRX de PDLLA présentent 4 pics
caractéristiques, dont deux pics sont très intences correspondant
aux distances interrétéculaires suivantes : 5,3 et 4,7 avec des
déviations 2è de 16,6° et 19,1°, respectivent. En plus,
deux pics de faibles intensités sont observées aux distances
rétéculaires suivantes : 6,0 et 4,0 avec des déviatios
2è de 14,7° et 22,3°, respectivement. Ces résultats
sont en accord avec ceux trouvés par Davide Brizzolara et
al. (D. Brizzolara, 1996).
Intensite

14,7°
16,6°
19,1°
22,3°
(b)
(d)
(a)
5 10 15 20 25 30 35 40 45 50
2 Thêta (°)
Figure 63. Les spectres DRX de poly(D,L-acide
lactique) : (a) PDLLA 1000, (b) PDLLA 3000 et (c) PDLLA
9000 Da.
Partie II. Caractérisation du principe actif
(Ibuprofène pur)
II.1. Analyse thermique
II.1.1. Etude TG/DTG
La figure 64 montre les courbes TG/DTG obtenues à 0,5
°C/min sous atmosphère d'azote pour l'ibuprofène pur. Les
courbes ATG-ATD de l'ibuprofène montrent une stabilité thermique
jusqu'à 125 °C. La décomposition thermique s'effectue entre
125 et 264 °C avec une perte de masse totale correspondante au pic
endothermique à 237,2 °C, ces résultats sont en accord avec
la littérature (G. Bannach, 2010).

dm/dt (%/min)
-10
-15
-20
-5
0
5
0
0 50 100 150 200 250 300 350 400 450 500
40
20
60
80
120
100
m (%)
TG
DTG
Température (°C)
Figure 64. Courbe de TG/DTG obtenu à 10
°C/min sous atmosphère d'azote pour l'ibuprofène pur.
II.1.2. Analyse DSC
La courbe DSC de l'ibuprofène pur est donnée
dans la figure 65. D'après cette dernière, l'ibuprofène
montre un pic endothermique correspondant à son processus de fusion
à 76,7 °C avec un enthalpie de fusion ÄHf de 61,29 J/g, ce
résultat est en accord avec celui trouvé par Juliana Bidone et
al. (J. Bidone, 2009).

Endotherme
Tf = 76,69 °C
0,2
0,0
-0,2
Flux de chaleur (mW.mg-1)
-0,4
-0,6
-0,8
-1,0
-1,2
-1,4
-1,6
-1,8
20 40 60 80 100 120
Température (°C)
Figure 65. Courbe DSC de l'ibuprofène
pur.
II.2. Analyse structural
II.2.1. Spectre IR-TF de l'ibuprofène
La figure 66 montre le spectre Infrarouge à
Transformé de Fourier (IR-TF) de l'ibuprofène pur. La comparaison
avec les spectres de l'ibuprofène trouvés dans la
litérature (J. Namur, 2009 ; G. Bannach, 2010), nous a
permet d'indexer notre spectre, et le tableau 18 suivant résume les pics
caractéristiques de l'ibuprofène pur ainsi que les types de
vibration des liaisons correspondates.
Tableau 18. Les bandes d'absorption infrarouge
caractéristiques de l'ibuprofène pur.
|
Nombre d'onde í (cm-1)
|
Liaison et type de vibration
|
|
3090
|
Vibration d'élongation de C-H aromatique
|
|
2955
|
Vibration d'élongation antisymétrique de CH3
|
|
1720
|
Vibration d'élongation de C=O (COOH)
|
|
1509
|
Vibration d'élongation de C-C cyclique
|
|
1420
|
Vibration élongation/déformation
antisymétrique de C-C-O-H
|
|
1269
|
Vibration d'élongation de C-O (COOH) et vibration de
|
|
1230
|
déformation de O-H
|
|
1184
|
|
|
935
|
Vibration de déformation hors du plan de O-H
(dimère acide)
|

3090
935
1420
2955
1269
1230
Transmittance (%)
1184
1720
4000 3500 3000 2500 2000 1500 1000 500
Nombre d'onde (cm-1)
Figure 66. Spectre IRTF de l'ibuprofène
pur.
II.2.2. Spectre DRX de l'ibuprofène
La diffraction des rayons X de l'échantillon
ibuprofène pur est donnée dans la figure 67. Le spectre DRX de
l'ibuprofène a révéle des reflexions de fortes
intensités et elles correspond aux distances
interétéculaires suivantes : 14,4, 7,2, 5,3, 4,4 et 4,0 avec des
pics caractéristiques à 6,1°, 12,3°, 16,6°,
20,2°, et 22,4°, respectivement, et ces résultats sont
comparables à ceux trouvés dans la litérature (J.
Bidone, 2009 ; N. V. Phadnis, 1997 ; A. FernándezCarballido, 2004 ; C.
Acquah, 2009).

3000
2500
2000
1500
1000
500
0
5 10 15 20 25 30 35 40 45 50
2 Thêta (°)
Intensite
16,632°
20,221°
22,390°
6,139°
12,257°
Figure 67. Spectre DRX de l'ibuproféne
pur.
Partie III. Etude et caractérisation des
différents mélanges PDLLA/IB
III.1. Microscopie électronique à balayage
(MEB)
La figure 68 montre la morphologie externe des microgranules
de l'ibuprofène (a), de poly(D,L-acide lactique) (b) et des
différents mélanges PDLLA/IB (mélanges physiques (c),
mélanges de fusion à chaud (d) et mélanges par
évaporation de solvant (e)). D'après ces images MEB,
l'ibuprofène (a) est apparu comme de grosses particules
légérement laisses en surface, et le poly(D,L-acide lactique) (b)
est apparu comme de petites particules. Les images MEB des différents
mélanges PDLLA/IB ((c), (d) et (e)) ont montré des particules qui
sont très différentes de celles des composés purs ; ces
particules sont apparu comme des masses homogènes froissées en
surface et de ptites tailles. En effet, la changement dans la morphologie des
particules formant les différents mélanges peut être le
résultat de l'enrobage des particules de l'ibuprofène par celles
de poly(D,L-acide lactique). Autrement dit, les images MEB ((c), (d) et (e))
ont montrée de grosses particules (ibuprofène) avec de petites
particules (poly(D,L-acide lactique)) adhérées à leurs
surfaces.

c d
e
Figure 68. Micrographies électronique
à balayage : (a) Ibuprofène, (b) Poly(D,L-acide lactique), (c)
Mélanges
a
b
physiques, (d) Mélanges en fusion et (e) Mélanges
par évaporation de solvant.
III.2. Spectroscopie infrarouge à transformé
de fourrier (IR-TF)
La méthode IR-TF a été choisie pour
caractériser les différents groupes fonctionnels de l'IB et de
PDLLA d'une part, et d'autre part pour étudier l'existence d'une
interaction (physique ou chimique) entre l'IB et le PDLLA. Les spectres IR-TF
de l'ibuprofène, poly(D,L-acide lactique) ainsi que leurs
mélanges binaires sont montrés dans les figures 69, 70 et 71 ;
chacune des trois figures montre la comparaison entre les spectres IR-TF des
produits brutes : IB (figures 69(a), 70(a) et 71(a)), PDLLA3000 (figures 69(b),
70(b) et 71(b)) et les spectres IR-TF des différentes formulations (IB +
x% PDLLA3000) de chacun des mélanges préparés.
La comparaison du spectre IR-TF de l'ibuprofène pur
avec les spectres des différentes formulations préparées,
nous a permet de remarquer que tous les pics caractéristiques de
l'ibuprofène pur sont présents dans les spectres IR-TF des
mélanges IB/PDLLA. Ce ci indique que la structure moléculaire de
la molécule d'ibuprofène a resté intact dans tous les
mélanges.
Transmittance (%)
2
4
0
3
1

(d)
(a)
(c)
(e)
(e)
4000 3500 3000 2500 2000 1500 1000 500
Nombre d'onde (cm-1)
Figure 69. Spectre IRTF des produits purs (a)
ibuprofène pur, (b) PDLLA 3000 pur, et des mélanges
physiques
(c) IB+5%PDLLA, (d) IB+15%PDLLA et (e) IB+25%PDLLA.
Chapitre IV. Résultats et discussion

4000 3500 3000 2500 2000 1500 1000 500
0
(a)
(b)
1
(c)
Transmittance (%)
(d)
2
(e)
3
4
Nombre d'onde (cm-1)
Figure 70. Spectre IRTF des produits purs (a)
ibuprofène pur, (b) PDLLA 3000 pur, et des mélanges à
fusion (c)
IB+5%PDLLA, (d) IB+15%PDLLA et (e) IB+25%PDLLA.
4000 3500 3000 2500 2000 1500 1000 500

Transmittance (%)
(d)
(f)
(a)
(c)
(e)
Nombre d'onde (cm-1)
Figure 71. Spectre IRTF des produits purs (a)
ibuprofène pur, (b) PDLLA 3000 pur, et des mélanges
par
évaporation de solvant (c) IB+5%PDLLA, (d) IB+15%PDLLA et (e)
IB+25%PDLLA.
Le spectre IR-TF de l'ibuprofène pur présente
une bande d'absorption distincte et très intense située dans la
région de l'absorption des carbonyles acides (í(C=O)) entre 1650
et 1800 cm-1 et précisément à 1720
cm-1 (figure 72(a)). Cette dernière peut être
utilisée pour identifier le groupe fonctionnel (C=O) de
l'ibuprofène d'une part, et pour identifier tous les changements
possibles dans la structure moléculaire de l'ibuprofène lorsqu'il
est mélanger ou recristalliser en présence de poly(D,L-acide
lactique), d'autre part. En effet, d'après les figures 72(A), 72(B) et
72(C) nous avons remarqué que le pic intense situé à 1720
cm-1 dans le spectre IR-TF de l'IB pur a été
décalé vers des valeurs supérieurs des nombres d'ondes
(í), cette même remarque a été rapportée dans
la littérature (F. Cilurzo, 2005 ; T. Phromsopha, 2009 ; S. G.
Kazarian, 2002 ; S. Nakayama, 2009), et ce là dans touts les
mélanges et dans toutes les formulations préparés sauf la
formulation F5 (IB + 5% PDLLA3000), ce qui ne peut être expliquer que par
la faible quantité de PDLLA (5%) incorporée entre les grains de
l'ibuprofène solide. Le décalage observé dans la
région des carbonyles varier d'une formulation à l'autre et d'un
mélange à l'autre. En effet, plus la quantité de PDLLA
incorporée dans l'IB est importante plus le décalage est
important, et plus le contact entre les molécules d'IB et celles de
PDLLA est important plus le décalage est important. Pour le
mélange physique le décalage varier de 2 cm-1 (F15)
à 4 cm-1 (F25), pour le mélange par fusion à
chaux le décalage varier de 4 cm-1 (F15) à 8
cm-1 (F25) et pour la mélange par évaporation de
solvant le décalage varier de 2 cm-1 (F15) à 6
cm-1 (F25), comme le montre bien la figure 72.
(A) (B)
2000 1900 1800 1700 1600 1500 2000 1900 1800 1700 1600 1500
Nombre d'onde (cm-') (C) Nombre d'onde
(cm-')

(a)
(b)
(d)
(c)
(e)
1759
1726 1722 1720
1720
2000 1900 1800 1700 1600 1500
Transmittance (%)

Transmittance (%)
Transmittance (%)
(a)
(d)
(c)
(e)
(b)
1759
1728 1724 1720
1720
(d)
(b)
(c)
(e)
(a)
1759
1724 1722
1720
1720
Nombre d'onde (cm-')
Figure 72. Spectre IR-TF dans la région
spectrale du carbonyle. (A) Mélanges physiques, (B) Mélanges
à fusion
et (C) Mélanges par évaporation de solvant de
IB et PDLLA3000.
Intensite

(1) IB pur
DPLLA3000 F25
6,139
6,162

(2) IB pur
PDLLA3000 F25
12,257
12,261
III.3. Analyse cristallographique des différents
échantillions
L'étude de la cristallinité de
l'ibuprofène dans les différents échantillions IB/PDLLA a
été éffectuée par la méthode de diffraction
des rayons X sur poudre (DRX). La diffraction des rayons X sur poudre est une
méthode très utilisée pour déterminer si un couple
de cristaux d'un principe actif donné sont polymorphes. En
général pour deux formes de cristaux, lorsque les positions des
pics sont identiques, ont dit que les deux cristaux possèdent la
même structure interne ; si les positions des pics sont
différentes, ont dit que les deux cristaux ont des structures
différentes et sont polymorphes (A. T. Karunanithi,
2007).
Les figures 73, 75 et 77 montrent les spectres DRX de
l'ibuprofène pur (a), de poly(D,Lacide lactique) (b) et des
différents mélanges IB/PDLLA (c), (d) et (e). D'après les
trois figures, il est clair que les pics caractéristiques du spectre DRX
de l'ibuprofène pur à 5-50° (2è) (figures 73a,75a et
77a) sont visibles dans les spectres DRX des différents mélanges,
ce qui indique la présence de l'ibuprofène sous sa forme
cristalline dans ces derniers.
Dans le mélange physique simple IB/PDLLA, les
structures cristallines des deux composés reste intact et que toutes les
fromulations ont montrées des spectres DRX avec approximativement les
mêmes positions (2è) des pics (figure 74). Donc, la
présence de différents polymorphes de l'IB dans ces
échantillons a été éliminée. Cependant, nous
avons constaté une légére variation dans les
intensités et largeurs des pics des différents formulations.
Intensite

Mélanges physiques
(e) (d) (c) (b) (a)
5 10 15 20 25 30 35 40 45 50
2 Thêta (°)
Figure 73. Diffraction des rayons X sur poudre
de (a) PDLLA3000, (b) IB pur, (c) IB+5%PDLLA3000 (F5), (d)
IB+15%PDLLA3000
(F15) et (e) IB+25%PDLLA3000 (F25).
5,0 5,2 5,4 5,6 5,8 6,0 6,2 6,4 6,6 6,8 7,0 11,6 11,8 12,0 12,2
12,4 12,6 12,8 13,0
Intensite

(3) IB pur
PDLLA3000 F25
16,632
16,642

(4) IB pur
PDLLA3000 F25
20,221
20,210
22,390
22,393
16,0 16,2 16,4 16,6 16,8 17,0 17,2 19,5 20,0 20,5 21,0 21,5 22,0
22,5 23,0
2 Thêta (°) 2 Thêta (°)
Figure 74. Diffraction des rayons X sur poudre
des produits purs (IB et PDLLA3000) et de la formulation F25
(mélange
physique) dans les intervalles des angles 2è de (1)
5,0°-7,0°, (2) 11,5°-13,0°, (3)
16,0°-17,2° et (4)
19,5°-23,0°.
Dans le mélange de fusion à chaud de l'IB et
PDLLA, les spectres DRX des différéntes formulations ont
indiqué la présence de l'ibuprofène cristallin mais avec
une petite augmentation dans les intensités relatives et dans les
largeurs des pics, d'une part, et avec une petite déviation des pics
vers des valeurs 2è inférieures, d'autre part.
5 10 15 20 25 30 35 40 45 50
2 Thêta (°)

Intensite
Mélanges de fusion à chaud
(e)
(d)
(c)
(b)
(a)
Figure 75. Diffraction des rayons X sur poudre
de (a) PDLLA3000, (b) IB pur, (c) IB+5%PDLLA3000 (F5), (d)
IB+15%PDLLA3000
(F15) et (e) IB+25%PDLLA3000 (F25).
Intensite

(1) IB
PDLLA3000 F25
6,063
6,139
(2) IB
PDLLA3000 F25
12,175 12,257
5,0 5,2 5,4 5,6 5,8 6,0 6,2 6,4 6,6 6,8 7,0 11,6 11,8 12,0 12,2
12,4 12,6 12,8 13,0
Intensite

(3) IB
PDLLA3000 F25
16,578
16,632
(4) IB
PDLLA3000 F25
20,164
20,221
22,078
22,390
16,0 16,2 16,4 16,6 16,8 17,0 17,2 19,5 20,0 20,5 21,0 21,5 22,0
22,5 23,0
2 Thêta (°) 2 Thêta (°)
Figure 76. Diffraction des rayons X sur poudre
des produits purs (IB et PDLLA3000) et de la formulation F25
(mélange
de fusion à chaud) dans les intervalles des angles 2è de (1)
5,0°-7,0°, (2) 11,5°-13,0°, (3)
16,0°-17,2°
et (4) 19,5°-23,0°.
Dans le cas du mélange par évaporation de solvant,
les spectres DRX des différéntes formulations ont aussi
indiqués la présence de l'ibuprofène cristallin mais avec
une petite augmentation dans les intensités relatives et dans les
largeurs des pics, d'une part, et avec une petite déviation des pics
vers des valeurs de 2è inférieures, d'autre part.
IntensiO

Mélanges par évaporation de solvant
(e)
(d)
(c)
(b) (a)
5 10 15 20 25 30 35 40 45 50
2 Thêta (°)
Figure 77. Diffraction des rayons X sur poudre
de (a) PDLLA3000, (b) IB pur, (c) IB+5%PDLLA3000 (F5), (d)
IB+15%PDLLA3000
(F15) et (e) IB+25%PDLLA3000 (F25).
hgensite

(1) IB
PDLLA3000 F25
4070 6,139
(2) IB
PDLLA3000 F25
12,159 12,257
5,0 5,2 5,4 5,6 5,8 6,0 6,2 6,4 6,6 6,8 7,0 11,6 11,8 12,0 12,2
12,4 12,6 12,8 13,0
hgensite

(3) IB
PDLLA3000 F25
14572 14632
(4) IB
PDLLA3000 F25
20,143
7n791
22,294
22,390
16,0 16,2 16,4 16,6 16,8 17,0 17,2 19,5 20,0 20,5 21,0 21,5 22,0
22,5 23,0
2 Thêta (°) 2 Thêta (°)
Figure 78. Diffraction des rayons X sur poudre
des produits purs (IB et PDLLA3000) et de la formulation F25
(mélange
par évaporation de solvant) dans les intervalles des angles 2è de
(1) 5,0°-7,0°, (2) 11,5°-13,0°,
(3)
16,0°-17,2° et (4) 19,5°-23,0°.
Nous avons constaté les mêmes remarques, que
celles observées pour la formulation F25, pour les autres formulations
(F5 et F15) dans les trois mélanges. Le tableau 19 résume les
valeurs de 2è et les intensités T correspondantes aux pics
caractéristiques de l'ibuprofène.
Tableau 19. Diffraction des rayons X des
différentes fomulations de l'ibuprofène avec le PDLLA3000.
|
Mélanges physiques
|
|
|
Echantillon
|
|
2è (°)
|
Intensité I (cps)
|
|
TB + 5%PDLLA3000
|
|
6,160 - 12,272 - 16,686 -
|
100,2214
|
- 47,0183 -
|
|
|
20,267 - 22,417
|
370,3842
|
- 330,0737 -
|
|
|
|
285,8180
|
|
|
TB + 15%PDLLA3000
|
|
6,133 - 12,260 - 16,641 -
|
121,8539
|
- 39,9801 -
|
|
|
20,213 - 22,363
|
407,2002
|
- 274,1543 -
|
|
|
|
241,9440
|
|
|
Mélanges de fusion à chaud
|
|
|
TB + 5%PDLLA3000
|
|
6,004 - 12,057 - 16,431 -
|
199,1964
|
- 74,1898 -
|
|
|
19,933 - 21,938
|
623,1784
|
- 516,0129 -
|
|
|
|
384,3003
|
|
|
TB + 15%PDLLA3000
|
|
6,022 - 12,166 - 16,497 -
|
129,0107
|
- 44,1353 -
|
|
|
20,026 - 22,244
|
532,0245
|
- 425,7441 -
|
|
|
|
212,9239
|
|
|
Mélanges par évaporation de
solvant
|
|
|
TB + 5%PDLLA3000
|
|
6,030 - 12,148 - 16,522 -
|
348,4015
|
- 105,5071 -
|
|
|
20,095 - 22,264
|
703,5086
|
- 541,9090 -
|
|
|
|
412,4559
|
|
|
TB + 15%PDLLA3000
|
|
6,032 - 12,115 - 16,543 -
|
202,0589
|
- 87,0501 -
|
|
|
20,080 - 22,217
|
606,3016
|
- 464,0741 -
|
|
|
|
410,3376
|
|
D'après les figures 74, 76 et 78 et les
résultats donnés dans le tableau 19, nous avons constaté
que les intensités relatives et la largeur des pics ont
légèrement augmentés comme c'est motionner ci-dessus, ce
ci peut être attribué a l'altération de la
morphologié des cristaux d'TB dans les mélanges (A. T.
Karunanithi, 2007). Par conséquent, l'abondance relative des
plans exposés à la source des rayons X aurait été
changée, produisant des variations dans les intensités relatives
des pics, ou ce ci peut être due à la variation dans la taille des
cristaux (H.
A. Carebani, 2001 ; M. Maneghini, 2008).
À la différence des spectres DRX des
mélanges physiques, où les positions des pics 2è ont
restées partiquement inchangées, les spectres DRX des autres
mélanges (mélanges de fusion à chaud et mélanges
par évaporation de solvant) ont montré, en plus de petites
variations dans les intensités et des largeurs des pics, de petites
variations dans les positions des pics vers des valeurs inférieures de
2è. Par consiquent, la présence de différents polymorphes
d'ibuprofène dans ces mélanges ne peut être exclu. En
effet, d'après Gul Madjid Khan (G. M. Khan, 2001), une
différence significative dans les spectres DRX doit être
prévu si un complex d'iclusion est formé, car la structure
cristalline sera changée. En plus, certains changemants comme positions
des pics, intensités des pics, et de petites différences dans les
distances interrétéculaires (d) indiquent la possibilité
d'avoir des intéractions entres le principe actif et le
polymère.
III.4. Tests de dissolution in vitro de
l'ibuprofène
Dans cette dernière partie, notre objectif est
d'étudier les profiles de libération de l'ibuprofène
à partir des microgranules de poly(D,L-acide lactique).
Dans un premier temps, nous avons étudié l'effet
du taux d'enrobage sur la vitesse de libération de l'IB à partir
de la matrice PDLLA. Puis, et pour une formulation donnée, nous avons
intéressés à l'étude de l'évolution des
profiles de libération de l'IB à patir de la matrice PDLLA
lorsque ce dernier est utilisé avec des masses moléculaires
différentes. Comme dans le coprs humain, en particulier dans la partie
gastrointestinale (GI), un médicament lorsqu'il est avalé passe
à travers plusieurs parties à des pH différents, donc il
est interissant de prendre en consédiration ce paramètre dans
notre étude. Effictivement, par la suite nous avons étudié
l'effet du pH du milieu de dissolution sur la vitesse de libération de
l'ibuprofène. Enfin, comme chacune des formulations a été
préparée par trois méthodes diffrentes, nous avons
étudié l'inffluence de la méthode de préparation
sur la vitesse de libération de l'IB à partir de la matrice
poly(D,L-acide lactique).
Les profiles de libération de l'ibuprofène a
partir de la matrice PDLLA ont été mesurés en utilisant la
spectroscopie UV-visible. Une courbe d'étalonnage de la concentration de
l'IB (C) comme une fonction de l'absorption (A) a été
utilisée pour calculer la masse de l'IB qui a été
libérée à partir des formulations. Le pourcentage de l'IB
libéré à partir des formulations à un temps
donné (t) a été calculer par le rapport entre la masse
libérée au temps t (Mt) et la masse de l'IB utilisée
initialement (M8) que multiplier par 100 :
X (%) = Mt / M8 * 100 (4)
III.4.1. Effet du taux d'enrobage
Les figures 79 et 80 montrent l'évolution du
pourcentage de l'IB libéré en fonction du temps à partir
de microgranules enrobés à différents taux d'enrobage (5,
15 et 25 %). Les profiles de libération de l'ibuprofène de la
figure 79 ont été obtenus à pH 7,4 (milieu alcalin),
tandis que ceux de la figure 80 ont été obtenus à pH 5,8
(milieu acide). Nous avons remarqué, d'après les deux figures,
que toutes les compositions sous formes de comprimées permettent
d'obtenir un profil de libération biphasique de type rapide/lent (ou
quick/slow), avec une phase initiale de libération rapide et importante
(ou « burst ») de l'ibuprofène faiblement associé
à la surface des microgranules de PDLLA, permettant d'obtenir une dose
initialement importante, suivie d'une phase de libération plus lente de
l'ibuprofène sur une durée plus longue. La figure 79 suivante
montre qu'à pH 7,4 entre 40 et 70% de la quantité d'IB initiale a
été libérée duarant les 60 premières minutes
(c'est la phase de libération rapide), suivi par une phase de
libération lente de la quantité d'IB restante sur une
période allon de 60 à 250 min. De même à pH 5,8
(figure 80), nous avons remarqué que durant la phase de
libération rapide environ 20 à 60% de l'IB initial a
été libéré dans les 600 premières
minutés (10 h), tandis que le reste de l'IB a été
libéré sur un intervalle temps plus large allon de 600 à
plus de 3000 minutes.
Chapitre IV. Résultats et discussion

0 20 40 60 80 100 120 140 160 180 200
Temps (min)
0 20 40 60 80 100 120 140 160 180 200
Temps (min)
0 20 40 60 80 100 120 140 160 180 200 220 240
100
Ibuprofen libere (%)
80
60
40
20
0
100
80
Ibuprofen libere (%)
60
40
20
0
(c)
100
Ibuprofene libere (%)
80
60
IB
40
F5 F15 F25
20
0
(a)
IB
F5 F15 F25
(b)
IB
F5 F15 F25
Temps (min)
Figure 79. Effet du taux d'enrobage sur les
cinétiques de libération de l'IB à partir des
microgranules de PDLLA
à pH 7,4. (a) mélanges physiques, (b)
mélanges en fusion et (c) mélanges par évaporation de
solvant.
Aux différents pH du milieu (7,4 et 5,8) et aux
différents types de mélanges (mélanges physiques,
mélanges en fusion et mélanges par évaporation de solvant)
étudiés, les cinétiques de libération ralentissent
quand le taux d'enrobage devient de plus en plus important comme il est
indiqué par le sens des flèches déssinées sur les
figures 79 et 80, et ce résultat est en accord avec la litérature
(T. Phromsopha, 2009 ; C. J. Thompson, 2007 ; B. Devrim, 2006 ; R.
Pignatillo, 2002). En d'autre terme, la tendance
générale observée est la suivante : la vitesse de
libération du principe actif diminue quand la quantité du
polymère incorporé augmente, et ce quel que soit le type du
mélange et le pH du milieu. En effet, d'après Fude Cui et al.
(F. Cui, 2005), l'augmentation de la concentration du
polymère dans la formulation va générer une matrice
polymère dense, résultant des pores de petites tailles dans le
volume de la matrice, et par conséquent la structure da la matrice
polymère devienne plus tortueuse, ce qui va provoqué par la suite
une vitesse de libération du principe actif plus lente. Cette tendance
peut être attribuée au phénomène de
dissolution-diffusion de l'ibuprofène entre les microgranules du PDLLA.
Une étude à été faite sur la dégaradation
hydrolytique de PDLLA et l'un de ses copolymères (PDLLA-PVP-PDLLA), en
tampon phosphate de pH 7,4 à 37°C, par Lei Xiong et al. (L.
Xiong, 2009). Ces auteurs ont costatté qu'après 3
semaines d'incubation, le PDLLA n'a subi aucune dégradation. En effet,
dans notre étude la dégradation de la matrice polymère
(PDLLA) est relativement lente par rapport à la période de
libération de l'ibuprofène, ce qui permet de déduire que
la libération est contrôlée par des
phénomènes de dissolution-diffusion de l'ibuprofène dans
le milieu environnant, et non par suite de la dégradation de la matrice
polymère. Une explication similaire à été
suggérée par C. J. Thompson et al (C. J.
Thompson,
2007), sachant qu'à la différence
de notre cas ces auteurs ont utilisés des polymères de types
copolyesters (SH-L509 et SH-L510) sous formes de microsphères.
Le phénomène de dissolution-diffusion est un
ensemble de deux étapes consécutive, la première
étape consiste à la diffusion du solvant (solution tampon)
à l'intérieur de la matrice polymère, ce ci va
provoqué la dissolution du principe actif. Ensuite, en deuxième
étape, la solution du principe actif peut quitter la matrice
polymère vers le milieu environnant (B.
Devrim, 2006).
0 500 1000 1500 2000 2500 3000
Temps (min)
0 500 1000 1500 2000 2500 3000
Temps (min)
0 500 1000 1500 2000 2500 3000

Ibuprofen libere (%)
100
40
20
60
80
0
(a)
100
(b)
IB F5 F15 F25
Ibuprofen libere (%)
40
20
60
80
IB
F5 F15 F25
0

Ibuprofen libere (%)
100
40
20
60
80
0
(c)
IB
F5 F15 F25
Temps (min)
Figure 80. Effet du taux d'enrobage sur les
cinétiques de libération de l'IB à partir des
microgranules de PDLLA
à pH 5,8. (a) mélanges physiques, (b)
mélanges en fusion et (c) mélanges par évaporation de
solvant.
Nous venons de voir l'effet de la proportion de PDLLA sur la
vitesse de libération de l'IB. Cependant, on peut éventuellement
mettre à profit ce phénomène. Pour une masse
moléculaire moyenne donnée de PDLLA, on peut choisir la
concentration de ce dernier de façon à obtenir une vitesse de
libération appropriée. Par exemple, on choisira une proportion du
polymère suffisante pour que la quantité de l'IB
libérée, en tampon phosphate de pH 7,4 à 37 °C, au
bout d'une heure, soit au moins égale 30 % de la quantité
initiale de l'IB et soit inférieure à 60 % de l'IB initial.
III.4.2. Effet de la masse moléculaire de la
matrice PDLLA
Les figures 81 et 82 montrent la comparaison des
cinétiques de libération de l'ibuprofène à partir
de microgranules enrobés en fonction de la masse moléculaire de
poly(D,L-acide lactique) utilisé pour une formulation donnée
(F15). Les différentes masses moléculaires comparées sont
1000, 3000 et 9000 g/mol. Ce test a été effectué dans deux
milieux de dissolution à deux valeurs de pH différentes, 7,4
(figure 81) et 5,8 (figure 82).
De même, les études de libération de l'IB
ont montré que la vitesse de libération de l'IB diminue lorsque
la masse moléculaire du PDLLA augmente, un résultat similaire a
été trouvé par C. J. Thompson et al. (C. J.
Thompson, 2007), et ce quel que soit le pH et le type du
mélange. Nous avons constaté que, dans le cas de la formulation
où la masse du PDLLA utilisée est de l'ordre de 1000, le profil
de libération est pratiquement superposé à celui de la
formulation qui contient PDLLA 3000. Ce résultat ne peut être
expliqué que par la faible différence entre les deux masses
moléculaires.
Pour expliquer l'effet de la masse moléculaire du
polymère sur la cinétique de libération d'un principe
actif, J. Bidone et al. (J. Bidone, 2009) et C. J. Thompson et
al. (C. J. Thompson, 2007) ont supposé que plus la
matrice polymère est hydrophile plus le milieu aqueux
pénètre facilement dans son volume interne, et par
conséquent la vitesse de dissolution du principe actif sera plus rapide.
En effet, d'après C. J. Thompson, l'hydrophilicité de la matrice
polymère (dans le cas des copolyesters) augmente avec l'augmentation du
nombre de groupes hydroxyles terminaux sur les chaînes de la matrice. Or,
dans le cas de poly(acide lactique) et ces dérivés (ex. PLGA),
plus la masse moléculaire de PLA diminue, plus le nombre de groupes
hydroxyles et carboxyles (groupes polaires) augmente (A.
Fernández-Carballido, 2004). Comme le milieu de dissolution
contient des molécules d'eau (milieu aqueux), et sachant que l'eau est
polaire, le poly(acide lactique) de faible masse et plus polaire peut
attiré les molécules d'eau en faisant des liaisons
hydrogènes avec eux.

(a)
F15 (PDLLA1000)
F15 (PDLLA3000)
F15 (PDLLA9000)
0 50 100 150 200 250 300
Temps (min)

(b)
F15 (PDLLA1000)
F15 (PDLLA3000)
F15 (PDLLA9000)
0 20 40 60 80 100 120 140 160 180 200
Temps (min)
0 50 100 150 200 250 300 350
110
100
90
80
70
60
50
40
30
20
Ibuprofene libere (%)
10
0
110
100
90
80
70
60
50
40
30
20
Ibuprofene libere (%)
10
0

Ibuprofene libere (%)
110
100
40
20
90
80
70
60
50
30
10
0
(c)
F15 (PDLLA1000)
F15 (PDLLA3000)
F15 (PDLLA9000)
Temps (min)
Figure 81. Effet de la masse
moléculaire de PDLLA sur les cinétiques de libération de
l'IB à pH 7,4. (a)
mélanges physiques, (b) mélanges en
fusion et (c) mélanges par évaporation de solvant.

110
100
90
Ibuprofen libere (%)
80
70
60
50
40
30
20
10
0
110
100
90
Ibuprofen libere (%)
80
70
60
50
40
30
20
10
0
(a)
(b)
F15 (PDLLA1000)
F15 (PDLLA3000)
F15 (PDLLA9000)
F15 (PDLLA1000)
F15 (PDLLA3000)
F15 (PDLLA9000)
0 500 1000 1500 2000 2500 3000
Temps (min) 0 500 1000 1500 2000 2500 3000
Temps (min)
Ibuprofen Mere (%)
110
100
40
20
90
70
60
50
30
80
10
0
(c)
F15 (PDLLA1000)
F15 (PDLLA3000)
F15 (PDLLA9000)


0 500 1000 1500 2000 2500 3000
Temps (min)
Figure 82. Effet de la masse
moléculaire de PDLLA sur les cinétiques de libération de
l'IB à pH 5,8. (a)
mélanges physiques, (b) mélanges en
fusion et (c) mélanges par évaporation de solvant.
De même, dans le cadre de l'exploitation des profiles de
libération obtenus ci-dessus, on peut déterminer facilement, par
de simples essais de routine, les concentrations du PDLLA dans les formulations
et les masses moléculaires du PDLLA, qui donneront les profiles
souhaités de libération de l'ibuprofène.
III.4.3. Effet du pH du milieu de dissolution
La figure 83 montre l'effet du pH du milieu de dissolution sur
la cinétique de libération de l'IB pur, et les figures 84, 85 et
86 montrent également l'effet du pH sur les cinétiques de
libération de l'IB à partir de microgranules enrobés
à différents taux d'enrobage (5, 15 et 25%). Les deux valeurs de
pH choisi sont 7,4 et 5,8.
La vitesse de libération de l'ibuprofène (figure
83) est plus grande dans la solution tampon avec une alcalinité plus
élevée. Autrement dit, la vitesse de libération l'IB
à pH 7,4 est plus grande que celle à pH 5,8. La même
constatation a été signalée lors de l'analyse des figures
84, 85 et 86, et ce quel que soit le taux d'enrobage et le type du
mélange. De tel comportement a été montré dans la
littérature (C. S. Proikakis, 2006 ; S. Davaran, 1996).
Pour expliquer se résultat, il est important de suivre le gonflement
d'un comprimé à base de PLA dans deux milieux aqueux de pH
différent, chose qu'ont pas faite. Cependant, C. S. Proikakis et al.
(C. S. Proikakis, 2003) ont étudié la
stabilité du poly(DL-acide lactique) dans des solutions aqueuses. Ces
auteurs ont observé que des comprimés en PLA immergés dans
un tampon (pH 7,4), absorbent environ dix fois plus que ceux immergés
dans un tampon (pH 5,4). En effet, cette différence a été
attribuée aux forces électrostatiques répulsives fortes
entre les anions
carboxylate (COO-) dérivés de la
dissociation des groupes carboxyliques terminaux à des pH

110
100
90
80
Ibuprotene Mere CY4
70
60
50
40
30
20
10
0
pH 7,4
pH 5,8
0 250 500 750 1000 1250 1500 1750 2000 2250 2500 2750 3000
3250
Temps (min)
Chapitre IV. Résultats et discussion
élevés. Les anions carboxylate sont responsable
des forces électrostatique répulsives fortes entre les
chaînes polymérique, ce qui mène à un degré
de gonflement important et à l'augmentation de la taille des pores dans
la matrice polymère, ce qui favorise une libération plus rapide
à pH 7,4.
En plus de sa, comme il est déjà mentionner dans
le chapitre I de la partie théorique (figure 21) la solubilité de
l'ibuprofène dépend du pH du milieu environnant. Dans une
solution acide ( 5,8) la solubilité de l'ibuprofène est moins de
1 mg/ml et augmente à des pH alcalin (environ 10 mg/ml).

0 500 1000 1500 2000 2500 3000
110
100
90
Ibuprofene libere (%)
80
70
60
50
40
30
20
10
0
(a)
pH 7,4
pH 5,8
Temps (min)
110
100
90
80
70
60
50
40
30
20
10
0
110
100
(b)
pH 7,4
pH 5,8
90
Ibuprofene libere (%)
80
70
60
50
40
30
20
10
0
0 500 1000 1500 2000 2500 3000
Temps (min) (c)
Ibuprofene libere (%)
pH 7,4
pH 5,8
0 500 1000 1500 2000 2500 3000
Temps (min)
Figure 84. Effet du pH sur la cinétique
de libération de l'IB à partir de la formulation F5 : (a)
mélange physique,
(b) mélange en fusion et (c) mélange
par évaporation de solvant.
- 109 -
Figure 83. Effet du pH sur la cinétique
de libération de l'ibuprofène pur.

0 500 1000 1500 2000 2500 3000
Temps (min)
0 500 1000 1500 2000 2500 3000
Temps (min)
110
100
90
Ibuprofen Mere (%)
80
70
60
50
40
30
20
10
0
110
100
90
Ibuprofene Mere (%)
80
70
60
50
40
30
20
10
0
pH 7,4
pH 5,8
(a)
pH 7,4
pH 5,8
(b)
110
100
90
Ibuprofene Mere (%)
80
70
60
50
40
30
20
10
0
pH 7,4
pH 5,8
(c)
Chapitre IV. Résultats et discussion
0 500 1000 1500 2000 2500 3000
Temps (min)
Figure 85. Effet du pH sur la cinétique
de libération de l'IB à partir de la formulation F15 : (a)
mélange
physique, (b) mélange en fusion et (c) mélange
par évaporation de solvant.
110
100
(a)
pH 7,4
pH 5,8
90
Ibuprofene libere (%)
80
70
60
50
40
30
20
10
0
0 500 1000 1500 2000 2500 3000
Temps (min)
110
100
90
80
70
60
50
40
30
20
10
0
0 500 1000 1500 2000 2500 3000
pH 7,4
pH 5,8
(b)
Temps (min)
Ibuprofene libere (%)
100
90
80
Ibuprofene libere (%)
70
60
50
40
30
20
10
0
0 500 1000 1500 2000 2500 3000
(c)
pH 7,4
pH 5,8
Temps (min)
Figure 86. Effet du pH sur la cinétique
de libération de l'IB à partir de la formulation F25 : (a)
mélange
physique, (b) mélange en fusion et (c) mélange
par évaporation de solvant.
III.4.4. Effet de la méthode de préparation
des mélanges
La figure 87 montre la comparaison des cinétiques de
libération de l'ibuprofène à partir de microgranules
enrobés en fonction du type du mélange utilisé pour un pH
donné. Les différents types de mélanges comparés
sont : mélanges physiques simples, mélanges de fusion à
chaud et mélanges par évaporation de solvant ; les taux
d'enrobages sont 5, 15 et 25 % et le pH utilisé est égale
à 7,4. Nous avons constaté que le type du mélange entre IB
et PDLLA influence d'une manière remarquable les cinétiques de
libération de l'ibuprofène avec un profil de libération
plus lent dans le cas du mélange par évaporation de solvant que
dans le cas des deux autres mélanges. Cette tendance est due
probablement à la différence entre les forces d'interaction
existantes entre l'IB et PDLLA dans chaque type de mélange. Il est
claire que les interactions entre l'ibuprofène et le poly(D,L-acide
lactique) sont meilleures lorsque ces derniers sont mélangés dans
un solvant approprié. Rosario Pignatello et al. (R. Pignatello,
2002), ont constaté que de meilleures interactions entre des
principes actifs de la famille AINS et des polymères de types Eudragit
ont été obtenues en solution (i.e. dans un solvant commun) et non
avec un simple mélange des deux composés.
IbuproRne lilbere (%)
110
100
40
20
90
70
60
50
30
80
10
0

F15
Mélange en fusion Mélange physique Mélange
par évaporation de solvant
0 25 50 75 100 125 150 175 200 225 250
0 25 50 75 100 125 150 175 200 225 250
0 50 100 150 200 250 300

F5
110
100
90
Ibuprofene lilbere (%)
80
70
60
50
40
30
20
10
0
Mélange en fusion Mélange physique Mélange
par évaporation de solvant

Temps (min) F25
110
100
90
80
70
60
50
40
30
20
10
0
Temps (min)
IbuproRne Mere (%)
Mélange en fusion Mélange physique Mélange
par évaporation de solvant
Temps (min)
Figure 87. Effet de la méthode
préparation des mélanges PDLLA/IB sur les profils de
libération de l'IB à partir
de microgranules enrobés
à différents taux d'enrobage (5, 15 et 25%) en tampon phosphate
de pH 7,4 à 37°C.
Il existe donc une tendance clair entre le type de
mélange utilisé et la vitesse de dissolution de
l'ibuprofène. Comparés à l'ibuprofène seul, les
mélanges de fusion à chaud ont donné un certain
ralentissement de la dissolution de l'ibuprofène, suivis par les
mélanges physiques et enfin par les mélanges par
évaporation de solvant. En effet, la variation dans les profils de
libération de l'IB est due probablement au degré de
miscibilité entre l'ibuprofène et PDLLA. Dans le cas des
mélanges physiques et de fusion à chaud, où le
mélange principe actif/ polymère s'effectue sans solvant, la
miscibilité entre les composés est incomplète
(T.
Vasconcelos, 2007). Or, la présence du
solvant peut influencé l'interaction principe actif/polymère,
l'orientation du principe actif sur la surface du polymère et la forme
cristalline du principe actif en présence du polymère. En plus,
le solvant induit le gonflement de la matrice polymère ce qui va
amélioré la diffusion du principe actif a l'intérieur de
la matrice polymère. Par conséquent, le principe actif se
disperse sur une surface plus large a l'intérieur de la matrice
polymère (A. C. Williams, 2005). Une étude a
été faite par A. Nokhodchi et al. (A. Nokhodchi, 2003)
sur la modification de l'état cristalline de la
phénytoïne en utilisant de différents solvants
(éthanol et acétone). Ces auteurs ont constaté que la
morphologie des cristaux de la phénytoïne a été
changée sous l'effet d'une interaction entre les différents faces
des cristaux et le solvant, et que cette interaction a été
assurée par des liaisons hydrogènes. Les résultats de
cette étude ont montré aussi que la vitesse de dissolution de la
phénytoïne recristallisée a été
inférieure a celle de la phénytoïne non traitée.
L'ibuprofène montre des différences dans la morphologie lorsqu'il
est recristallisé dans de divers solvants (alcool et hexane), et que ces
différences dans la morphologie des cristaux d'ibuprofène n'ont
pas été attribuées aux changements dans la structure
cristalline de l'ibuprofène (ou différents polymorphes) mais
c'est due seulement a des changements dans les vitesses de croissance des faces
spécifiques (A. T. Karunanithi, 2007).
III.5. Interactions ibuprofène/poly(D,L-acide
lactique)
La question qu'on doit poser dans ce cas est la suivante :
existe-elles des forces d'interactions entre le poly(D,L-acide lactique) et
l'ibuprofène a l'échelle moléculaire, qui seront peut
être responsables de la rétention des molécules d'IB entre
les molécules de PDLLA ?.
Pour répondre a cette question, plusieurs techniques de
caractérisation, a savoir MEB, IRTF et DRX ont été
exploitées. En plus, et pour mettre a profit cette étude, des
testes de dissolution ont été effectués pour
étudier d'une part l'existence et l'importance de cette interaction
PDLLA/IB, et d'autre part pour donner une idée sur la possibilité
d'utiliser la matrice PDLLA comme vecteur de médicaments.
La microscopie électronique a balayage (MEB) nous a
permet de voir la morphologie cristalline externe des différents
échantillons. Or, la morphologie cristalline influence plusieurs
paramètres de l'ingénierie pharmaceutique et de la biopharmacie
comme fluidité, compression, compressibilité, solubilité
et la dissolution du principe actif (M. Manish, 2005). Dans
notre étude, la comparaison des micrographies électroniques a
balayage des produits purs (PDLLA et IB) avec celles des différents
échantillons (figure 68) nous a révélé que le
contact entre l'IB et PDLLA a provoqué certains changements dans la
morphologie externe des microgranules, a savoir réduction de la taille
et rugosité de la surface des microgranules. La cristallinité des
microgranules d'IB enrobés par PDLLA a été
étudiée par la technique de diffraction des rayons X sur poudre.
Cette dernière est utilisée pour détecter directement les
propriétés cristallines d'un matériau. La comparaison du
spectre DRX de l'ibuprofène pur avec les spectres des différents
mélanges (figures 74, 76 et 78) nous a permet de voir certaines
différences a savoir augmentation de l'intensité des pics,
élargissement des pics et décalage des positions des pics vers
des valeurs inférieures de 2è. Souvent, le principe actif subi
une dispersion moléculaire dans la matrice polymérique et le
mélange résultant devient amorphe. Cependant, dans certains cas
comme la notre, le principe actif subi une dispersion particulaire a
l'intérieur de la matrice polymérique au lieu d'une dispersion
moléculaire, ce qui est due probablement a la faible solubilité
du principe actif dans la matrice polymérique (S. Freiberg,
2004). Pour détecter les changements possibles a
l'échelle moléculaire, les différents échantillons
ont été analysés par spectroscopie infrarouge a
transformé de fourrier (IR-TF). La figure 72 précédente
compare les spectres des produits brutes (IB et PDLLA) avec les
Chapitre IV. Résultats et discussion

O
(b) HO

O
(c) HO
OH
O
n
O
n
O
O
O
O
OH

O H
O
Figure 88. Les éventuelles interactions
dans les dispersions solides IB/PDLLA. (a) dimère symétrique
d'IB, (b)
PDLLA seul et (c) IB liée à la chaîne de PDLLA
par une liaison hydrogène.

O
H
O
O
H
O
(a)
spectres des différents échantillons dans la
région spectrale de la vibration d'élongation des carbonyles
(í(C=O)). Les changements dans la région spectrale des carbonyles
sont très importantes pour élucider l'état
moléculaire du principe actif. L'analyse des spectres IR-TF des
différents échantillons nous a révélé que la
bande de vibration d'élongation du groupe C=O de l'ibuprofène a
changée sa position vers la région des nombres d'ondes
supérieurs. Ces résultats indiquent la rupture des liaison
hydrogènes dans les dimères ibuprofèneibuprofène
(figure 88a), qui sont caractéristiques de la forme solide de
l'ibuprofène, une fois se dernier est incorporé dans la matrice
PDLLA. En effet, le fait que la bande d'absorption d'ibuprofène dans la
région spectrale des carbonyles a été
décalée vers des nombres d'ondes supérieurs, il est
évident qu'une interaction intermoléculaires entre
l'ibuprofène et PDLLA existe, et que cette interaction est
assurée par des liaisons hydrogènes entre le groupe carbonyle du
poly(D,L-acide lactique) et le groupe hydroxyle de l'ibuprofène comme il
est montré dans la figure 88c. l'existence des liaisons
hydrogènes entre certains polymères et l'ibuprofène a
été signalée par plusieurs auteurs, à savoir T.
Phromsopha et Y. Baimark (MPEG-b-PDLL/IB) (T. Phromsopha,
2009), S. G. Kazarian et al. (PVP/IB) (S. G. Kazarian,
2002), S. Nakayama et al. (HPMC/IB) (S. Nakayama,
2009), Svetla Bogdanova et al. (PVP/IB) (S. Bogdanova,
2005).
La libération contrôlée est une
caractéristique possible est souhaitable pour les systèmes de
vectorisation de principes actifs. Les facteurs influençant la vitesse
de libération du principe actif sont généralement la
structure et les propriétés chimiques associées à
la matrice polymère et au principe actif (S. Freiberg,
2004). Comme c'est déjà mentionner dans la partie
théorique (chapitre I) que l'ibuprofène est un
antipyrétique et analgésique de la famille des anti-inflammatoire
non-stéroïdien (AINS). Ce médicament est utilisé pour
le soulagement des douleurs et l'inflammation dans des conditions
dysménorrhée, migraine, douleurs postopératoires et
douleurs dentaires. Il est aussi utilisé dans les troubles chroniques
comme ankylose spondylarthrite, ostéoarthrite et rhumatisme articulaire.
En plus, l'ibuprofène est facilement absorbé dans la partie
gastro-intestinale (GI). Par conséquent, l'ibuprofène provoque
certaines irritations dans la muqueuse gastro-intestinale et possède un
goût âpre. Cependant, la demi-vie synoviale courte de
l'ibuprofène (2 h) exigerait des injections fréquentes pour
maintenir le niveau thérapeutique efficace au voisinage du site
d'action. Pour cette raison une libération prolongée de
l'ibuprofène est souhaitable (B. Devrim, 2006). Pour
obtenir une libération prolongée, il faut retenir le principe
actif à l'intérieur d'une matrice polymérique d'où
il sera progressivement libéré. Pour ce faire, plusieurs
techniques peuvent être utilisées. Parmi ces techniques, c'est
l'utilisation des matrices polymériques hydrophobes et
biodégradables, où on peut jouer sur la masse et la proportion de
ses polymères pour moduler la cinétique de libération du
principe actif. Concrètement, nous avons mélangé les
microgranules d'ibuprofène avec celles d'un polymère hydrophobe,
biodégradable et biorésorbable qui est le poly(D,L-acide
lactique) au travers duquel la diffusion du principe actif peut se faire. Comme
c'est déjà signalé dans le paragraphe III.4.1, la
diffusion de l'ibuprofène à travers la matrice PDLLA est
gouvernée par le gonflement de cette dernière dans le milieu
tampon et non pas par la dégradation de la matrice polymérique.
Nous avons peut exploité ce phénomène (i.e. gonflement de
la matrice polymérique) pour expliquer comment que la proportion et la
masse moléculaire du PDLLA incorporé dans la formulation peut
influé sur les cinétiques de libération de
l'ibuprofène. En plus, le gonflement de la matrice polymérique
est directement lié au pH du milieu tampon, tel que plus le pH est
important (milieu alcalin) plus le gonflement est important. En bref, et dans
tous les cas, la diffusion du principe actif sous l'effet du gonflement de la
matrice polymérique s'effectue comme suit : le fluide environnant
pénètre à l'intérieur de la matrice à
travers des micropores. Un fort gradient de pression osmotique, entre
l'intérieur de la matrice polymérique et l'environnement
extérieur, permet une importante entrée d'eau dans la matrice et
donc son gonflement. Cela à pour conséquence une dilatation des
pores, qui accentue la fuite du principe actif solubilisé vers le milieu
extérieur (J. Pommay, 2006).
Enfin, les interactions entre les groupes carbonyle de PDLLA
et les groupes hydroxyle de l'ibuprofène peuvent interrompre la
cristallisation de l'ibuprofène dispersé dans la matrice
polymère. Les vitesses de libération de l'ibuprofène
à partir de la matrice PDLLA peuvent être manipulées par la
variation de certains paramètres comme proportion du PDLLA dans la
formulation, masse moléculaire du PDLLA et pH du milieu environnant. Le
poly(D,L-acide lactique) est donc un candidat très intéressant
pour des applications dans le domaine de vectorisation de principes actifs.
CONCLUSION GÉNÉRALE
Conclusion générale
Depuis quelques années, une des équipes du
Laboratoire des Matériaux Organique (LMO) de l'Université
A/Mira-Bejaïa s'intéresse à l'élaboration de
polymères biodégradables à vocation biomédicale. Il
a ainsi acquis une expérience dans l'élaboration de
polymères à base de polyesters tel le poly(acide lactique) par la
méthode de polymérisation par polycondensation
azéotropique.
Ce travail de mémoire s'inscrit dans la
continuité des travaux de recherches menées au LMO. L'objectif de
ce travail est d'étudier l'effet du mode de préparation de
formulations sur les interactions entre l'ibuprofène et le
poly(D,L-acide lactique) qui est un polymère biodégradable
capable d'encapsuler, transporter et de libérer, certains
molécules insolubles dans les milieux aqueux, de manière
contrôlée en jouant sur différents paramètres (taux
d'enrobage, masse moléculaire, pH...).
Le premier objectif de ce travail consiste à
l'élaboration d'une matrice polymère biodégradable qui est
le poly(D,L-acide lactique) à différentes masses
moléculaires par polycondensation azéotropique sous une
atmosphère inerte. Nous avons obtenu trois polymères de masses
moléculaires différentes (1000, 3000 et 9000) Da.
Le deuxième objectif est préparer et
caractériser les différentes formulations entre le PDLLA et
l'ibuprofène afin d'étudier les interactions entre ces deux
composés. Nous avons utilisé trois méthodes de
préparations différentes (mélange physique simple,
mélange par fusion à chaud et mélange par
évaporation de solvant).
Cette étude indique que le contact entre
l'ibuprofène et le PDLLA a provoqué certains changements dans la
morphologie externe des microgranules, à savoir réduction de la
taille et rugosité de la surface des microgranules, comme il est
indiquer par les images MEB.
Le changement dans la morphologie des particules des
différents mélanges a été aussi
vérifié par diffraction des rayons X. En effet, l'analyse DRX
nous a permis de voir certaines différences à savoir,
augmentation de l'intensité des pics, élargissement des pics et
décalage des positions des pics vers des valeurs inférieures de
2è. Le décalage des positions des pics a été
observé dans le cas des mélanges par fusion à chaud et par
évaporation de solvant. Ce résultat indique dans ce cas que la
structure cristalline de l'ibuprofène a été
altérée (i.e. présence de différents polymorphes
d'IB). Les résultats DRX nous a révélé que
l'ibuprofène a subi une dispersion particulaire entre les grains du
PDLLA au lieu d'une dispersion moléculaire car une fois
mélangé avec le PDLLA, l'ibuprofène a conservé sa
structure cristalline. En plus, certains changements comme positions des pics,
intensités des pics indiquent la possibilité d'avoir des
interactions entres le principe actif et le polymère.
Afin de mieux comprendre interactions existantes entre les
molécules d'ibuprofène et celle de PDLLA, nous avons fait appel
la spectroscopie infrarouge (IRTF). Cette étude nous a
révélée que la bande d'absorption d'ibuprofène dans
la région spectrale des carbonyles a été
décalée vers des nombres d'ondes supérieurs. Donc, il est
évident qu'une interaction intermoléculaires entre
l'ibuprofène et PDLLA existe et elle est assurée par des liaisons
par pont hydrogènes entre le groupe carbonyle du poly(D,L-acide
lactique) et le groupe hydroxyle de l'ibuprofène.
Enfin, le dernier objectif est la détermination du
profil de libération pour chaque formulation par un test de dissolution
in vitro dans le but de suivre l'influence de différents facteurs,
notamment le pourcentage de polymère incorporé pour l'enrobage du
principe actif, le pH, la masse moléculaire du polymère, pouvant
affecter la vitesse de libération du principe actif. Cette étude
indique que toutes les formulations permettent d'avoir un profil de
libération biphasique de type rapide/lent (ou quick/slow), avec une
phase initiale de libération rapide et
importante (ou << burst >>) de l'ibuprofène
suivie d'une phase de libération plus lente sur une durée plus
longue.
Pour les deux pH du milieu (7,4 et 5,8) et les trois types de
mélanges (mélanges physiques, mélanges par fusion à
chaud et mélanges par évaporation de solvant)
étudiés, les cinétiques de libération ralentissent
quand le taux d'enrobage augmente (i.e. la quantité du PDLLA
incorporée dans la formulation). De même, les études de
libération ont montré que la vitesse de libération de l'IB
diminue lorsque la masse moléculaire du PDLLA augmente et ce pour les
deux pH et les trois type du mélange.
Le pH du milieu joue un rôle important dans la
détermination des profils de libération de l'ibuprofène.
En effet, la vitesse de libération de l'ibuprofène est plus
importante dans une solution tampon avec une alcalinité plus
élevée. La comparaison des profils de libération d'une
même formulation, préparée avec différentes
méthodes, nous a permis de constater que le type de mélange
influe d'une manière remarquable sur les cinétiques de
libération de l'ibuprofène. Selon les résultats obtenus,
les formulations préparées par la méthode
d'évaporation de solvant ont présenté les profils de
libération les plus lents.
Enfin, la morphologie, la taille et le profil de
libération des microgranules ont été tous affectées
par les paramètres de fabrication et les conditions environnementales
à savoir le rapport PDLLA/IB, la masse moléculaire du PDLLA, le
pH de la solution et la méthode de préparation des
mélanges.
Un avantage des formulations préparées dans ce
travail, on peut par le choix de la masse moléculaire de la matrice
polymère, de sa concentration et de la méthode de
préparation des formulations, il est possible d'adapter et de faire
varier les profils de libération de l'ibuprofène. Le
poly(D,L-acide lactique) est donc un candidat très intéressant
pour des applications dans le domaine de vectorisation de principes actifs.
Comme perspectives,
À partir de ce travail, d'autres axes d'études
peuvent être proposés afin de mieux comprendre l'utilité de
la matrice poly(acide lactique) comme vecteur de principes actifs. Tout
d'abord, du point de vue synthèse, il serait intéressant
d'utiliser la méthode de synthèse
par ouverture du cycle du lactide afin d'aboutir à des
masses moléculaires plus élevées quiseront peut
être plus adéquates pour une libération plus lente du
principe actif.
Du point de vue méthode de préparation des
mélanges, il serait intéressant d'une part d'optimiser les
paramètres opératoires, tels température, durée du
mélange, type du solvant (polaire, apolaire), vitesse d'agitation, etc.
D'autre part, il serait intéressant d'utiliser d'autres techniques
préparatoires telle la polymérisation in situ du monomère
en présence du principe actif pour mieux le retenir. En effet, cette
méthode peut provoquer des interactions entre le polymère et le
principe plus fortes assurées par des liaisons covalentes (exemple :
liaison ester).
Du point de vue structure et architecture du polymère,
il serait intéressant d'élaborer des copolymères à
blocs amphiphiles biodégradables. Ces derniers ont la capacité
à s'autoassocier pour former des structures de tailles
nanométriques et de morphologies variées couramment
appelés << micelles >>. En effet, de part leurs
propriétés plus améliorées (solubilité,
degré d'encapsulation ...) que celles des homopolymères, les
copolymères à blocs amphiphiles biodégradables peuvent
être plus adéquats pour une libération plus lente du
principe actif et peuvent être plus adaptés pour l'organisme
vivant.
Comme, les autorités de santé internationales
demandent aux développeurs de médicaments de garantir autant que
possible la stabilité des médicaments en présence d'alcool
dans le
milieu de dissolution. Il serait intéressant
d'évaluer la résistance à l'alcool des formulations
préparées.
Enfin, les études de dissolution
réalisées in vitro dans des conditions qui simulent les
conditions physiologiques possèdent une valeur prédictive de ce
qui se passe in vivo. A cet effet, à l'avenir nous envisageons d'aborder
des tests biologiques pour déterminer les facteurs influençant le
devenir in vivo des mélanges préparés. Malgré que
dans ce domaine, la route est longue, nous pouvant toujours travailler en
collaboration avec des biologistes et des pharmacologues pour assurer le bon
déroulement de ces évaluations biologiques.
RÉFÉRENCES BIBLIOGRAPHIQUES
Références bibliographiques
-A-
A. A. Freer, J. M. Buyan, N. Shankland, D. B. Sheen. Structure of
(S)-(+)-ibuprofen. Acta Cryst. C49 (1993) 1378-1380.
A. A. Shah, F. Hasan, A. Hameed, S. Ahmed. Biological degradation
of plastics: A comprehensive review. Biotechnology Advances 26 (2008)
246-265.
A. C. Williams, P. Timmins, M. Lu, R. T. Forbes. Disorder and
dissolution enhancement : Deposition of ibuprofen on to insoluble polymers.
European Journal of Pharmaceutics Sciences 26 (2005) 288-294.
A. F. Kidonieus. Controlled-Release Technologies: Methods,
Theory, and applications, CRC Press, Inc., Boca Raton, FL, 213 (1987).
A. Fernández-Carballido, R. Herrero-Vanrell, I. T.
Molina-Martínez, P. Pastoriza. Biodegra dable ibuprofen-loaded PLGA
microspheres for intraarticular administration: Effect of Labrafil addition on
release in vitro. Inter J of Pharm 279 (2004) 33-41.
A. G. Andreopoulos, E. Httzi, M. Doxastakis. Synthesis and
properties of poly(lactic acid). Journal of Materials Science : Materials in
medicine 10 (1999) 29-33.
A. J. Romero a, C. T. Rhodes. Stereochemical aspects
of the molecular pharmaceutics of ibuprofen. J. Pharm. Pharmacol., 45 (1993)
258.
A. J. Romero b, L. Savastano, C. T. Rhodes. Monitoring
crystal modifications in systems containing ibuprofen. Int. J. of Pharm., 99
(1993) 125.
A. K. Bajpai, S. K. Shukla, S. Bhanu, S. Kankane. Responsive
polymers in controlled drug delivery. Progress in Polymer Science 33 (2008)
1088-1118.
A. L. Andrady. (Biodegradability of polymers) physical properties
of polymers handbook. (Ed.) Springer, New York (2007).
A. M. Deboeck, P. Maes, P. Baudier. Eutectic mixtures for oral
administration contg. Non steroid anti-inflammatory agent and fatty acid
glyceride(s). Patent, EP- 349509 (1990).
A. M. Hillery, A. W. Lloyd, J. Swarbrick. Drug delivery and
targeting for pharmacists and pharmaceutical scientists. Ed. Taylor &
Francis (2005).
A. Nalbandi. Kinetics of thermal degradation of polylactic acid
under N2 atmosphere. Iranian Polymer Journal 6 (2001), 371-376.
A. Nokhodchi, N. Bolourtchian, R. Dinarvand. Crystal modification
of phenytoin using different solvents and crystallization conditions.
International Journal of Pharmaceutics 250 (2003) 85-97.
A. P. Gupta, V. Kumar. New emerging trends in synthetic
biodegradable polymers- Polylactide: A critique. European Polymer Journal 43
(2007) 4053-4074.
A. S. Hoffman. The origins and evolution of
«controlled» drug delivery systems. Journal of Controlled Release 132
(2008) 153-163.
A. T. Karunanithi, C. Acquah, L. E. K. Achenie, S.
Sithambaram, S. L. Suib, R. Gani. An experimental verification of morphology of
ibuprofen crystals from CAMD designed solvent. Chemical Engineering Science 62
(2007) 3276 - 3281.
A. V. Kabanov, E. V. Batrakova. Polymer Nanomaterials . T. Ikezu
and H. E. Gendelman (ed.), Neuroimmune Pharmacology (2008).
A. Wawrezinieck, J.-M. Péan, P. Wüthrich, J.-P.
Benoit. Biodisponibilité et vecteurs particulaires pour la voie orale.
Médecine/Science. 24 (2008) 659-664.
-B-
B. A. Chabner. Clinical strategies for cancer treatment: the
role of drugs. Cancer chemotherapy and biotherapy: principles and practice. B.
A. Chabner and D. L. Longo. Philadelphia, Lippincott Williams & Wilkins
(2006) 1-14.
B. Devrim, K. Canefe. Preparation and evaluation of modified
release ibuprofen microspheres with acrylic polymers (Eudragit®) by
quasi-emulsion solvent diffusion method: Effect of variables. Acta Poloniae
Pharmaceutica - Drug Research, 63(6) (2006) 521-534.
B. Gupta, N. Revagade, J. Hilborn. Poly(lactic acid) fiber : An
overview. Prog. Polym. Sci. 32 (2007) 455-482.
B. Jiang, L. Hu, C. Gao, J. Schen. Ibuprofen-loaded
nanoparticles prepared by coprecipitation method and their release properties.
International Journal of Pharmaceutics 304 (2005) 220- 230.
B. Luppi, T. Cerchiara, F. Bigucci, I. Orienti, V. Zecchi.
pH-sensitive polymeric physicalmixture for possible site-specific delivery of
ibuprofen. European Journal of Pharmaceutics and Biopharmaceutics 55 (2003)
199-202.
B. M. Disher, D. A. Hammer, F. S. Bates, D. E. Disher. Polymer
vesicles in various media. Curr. Opin. Colloid Interface Sci. 5 (2000)
125-131.
-C-
C. Acquah, A. T. Karunanithi, M. Cagnetta, L. E. K. Achenie,
S. L. Suib. Linear models for prediction of ibuprofen crystal morphology based
on hydrogen bonding propensities. Fluid Phase Equilibria 277 (2009) 73-80.
C. Bastioli. Handbook of biodegradable polymers. Ed. Rapra
Technology Limited. United Kingdom (2005).
C. Chen, L. Dong, M. K. Cheung. Preparation and characterization
of biodegradable poly(Llactide)/chitosan blends. European Polymer Journal 41
(2005) 958-966.
C. De Brabander, C. Vervaet, L. Van Bartel, J.-P. Remon.
Bioavailability of ibuprofen from hot-melt extruded mini-matrices.
International Journal of Pharmaceutics 271 (2004) 77-84.
C. J. Thompson, D. Hansford, S. Higgins, C. Rostron, G. A.
Hutcheon, D. L. Munday. Evaluation of ibuprofen-loaded microspheres prepared
from novel copolyesters. International Journal of Pharmaceutics 329 (2007)
53-61.
C. K. Hebert, R. E. Williams, R. S. Levy, R. L. Barrack. Cost of
treating an infected total knee replacement. Clinical Orthopaedics and Related
Research 331 (1996) 140-5.
C. Kaprarissides, S. Alexandridou, K. Kottik, S. Chaitidous.
Recent advences in novel drug delivery systems. Journal of Nonotechnology
Online DOI: 10.2240/azojona0111 (2006),
http://www.azonano.com/oras.asp.
C. M. Boutry, R. Kiran, F. Umbrecht, C. Hierold. Processing
and quantitative analysis of biodegradable polymers (PLLA and PCL) thermal
bonding. J. Micromech. Microeng. 20 (2010) 13.
C. M. Perry, R. N. Brogden, Goserelin. A review of its
pharmaceutic and pharmacokinetic properties, and therapeutic use in benign
gynecological disorders drugs, 51 (1996) 319-349.
C. S. Proikakis, N. J. Mamouzelos, P. A. Tarantili, A. G.
Andreopoulos. Stability of DL-poly(lactic acid) in aqueous solutions. J. Appl.
Polym. Sci. 87 (5) (2003) 795-804.
C. S. Proikakis, P. A. Tarantili, A. G. Adreopoulos. Synthesis
and characterization of low molecular weight polylactic acid. Journal of
Elastomers and Plastics, 34 (2002) 49-63.
C. S. Proikakis, P. A. Tarantili, A. G. Andreopoulos. The role
of polymer/drug interactions on the sustained release from poly(DL-lactic acid)
tablets. European Polymer Journal 42 (2006) 3269-3276.
C. Vautheir-Holtzscherer, S. Benabbou, G. Spen lehauer, P.
Couveur. Methodology for preparation of ultradispersed polymer systems. S. T.
P. Pharma 1(1991) 109-116.
-D-
D. Brizzolara, H.-J. Cantow, K. Diederichs, E. Keller, A. J.
Domb. Mechanism of the stereocomplex formation between enantiomeric
poly(lactide)s. Macromolecules 29 (1996) 191-197.
D. E. Henton, Patrick Gruber, Jim Lunt and Jed Randall.
Polylactic acid technology. 1741 (2005) 527-578.
D. F. Williams. Enzymatic hydrolysis of poly(lactic acid). Eng.
Med. 10 (1981) 5-7.
D. G. I. Kingston. History and chemistry. Paclitaxel in
cancer treatment. W. P. McGuire and
E. K. Rowinsky. New York, Marcel Dekker (1995) 1-34.
D. Garlotta. A Literature Review of Poly(Lactic Acid). Journal of
Polymers and the Environment, 9(2) (2001) 63-84.
D. Jones. Pharmaceutical Applications of Polymers for Drug
Delivery. Rapra Technology Limite. United Kingdom (2004).
D. Muster. Médicament de l'inflammation. EMC-Stomatologie
1 (2005) 21-29.
D. R. Ryan, R. Garcia-Carbonero. Cytidine Analogues. Cancer
chemotherapy & biotherapy: Principles and practice. B. A. Chabner and D. L.
Longo. Philadelphia, Lippincott Williams & Wilkins (2006) 183-211.
-E-
E. Allémann, R. Gurny, E. Doelker. Drug-loaded
nanoparticles. Preparation methods and drug targeting issues. Eur. J. Pharm.
Biopharm. 39 (1993) 173-191.
E. Aurélie, F. Teddy, Polymères et
biodégradabilité, état des lieux et perspectives, France,
(2007) 1-32.
E. Autret-Leca. Anti-inflammatoires non
stéroïdiens et analgésie post opératoire : encore des
controverses. Centre Régional de Pharmacovigilance et d'Information sur
le médicament. France (2006).
E. Chiellini, H. Gil, G. Braunegg, J. Buchert, P. Gatenholm,
M. van der Zee. Biorelated polymers: Sustainable polymer science and
technology. Kluwer Academic/Plenum Publishers (2001).
E. K. Rowinsky. Antimicrotubule agents. Cancer chemotherapy
and biotherapy : principles and practice. B. A. Chabner and D. L. Longo.
Philadelphia, Lippincott Williams & Wilkins (2006) 237-282.
E. Mathiowitz. Encyclopedia of controlled drug delivery. Vols. I
& II. Wiley, New York (1999).
E. R. Gillies, J. M. J. Fréchet. Dendrimers and dendritic
polymers in drug delivery. DDT 10 (1) (2005) 35-43.
E. Rudnik. Compostable polymer materials. First edition, Elsevier
Ltd (2008).
E. T. H. Vink, K. R. Rábago, D. A. Glassner, P. R.
Gruber. Applications of life cycle assessment to NatureWorksTM polylactide
(PLA) production. Polymer Degradation and Stability 80 (2003) 403-419.
-F-
F. Aulenta, W. Hayes, S. Rannard. Dendrimers: a new class of
nanoscopic containers and delivery devices. Eur. Polym. J. 39 (2003)
1741-1771.
F. Cilurzo, P. Minghetti, A. Casiraghi, L. Tosi, S. Pagani, L.
Montanari. Polymethacrylates as crystallization inhibitors in monolayer
transdermal patches containing ibuprofen. European Journal of Pharmaceutics and
Biopharmaceutics 60 (2005) 61-66.
F. Cui, D. Cun, A. Tao, M. Yang, K. Shi, M. Zhao, Y. Guan.
Preparation and characterization of melittin-loaded poly (DL-lactic acid) or
poly (DL-lactic-co-glycolic acid) microspheres made by the double emulsion
method. Journal of Controlled Release 107 (2005) 310- 319.
F. H. Martini. Fundamentals of anatomy and physiology
(5th ed.), Prentice Hall, NJ, (2002) 847.
F. Meng, C. Humstra, G. H. Engbers, J. Feijen. Biodegradable
polymersomes. Macromolecules 36 (2003) 3004-3006.
F. Rancan, D. Papakostas, S. Hadam, S. Hackbarth, T. Delair,
C. Primard, B. Verrier, W. Sterry, U. Blume-Peytavi, A. Vogt. Investigation of
polylactic acid (PLA) nanoparticles as drug delivery systems for local
dermatotherapy. Pharmaceutical Research 26(8) (2009).
F. Schwark. Influence factors for scenario analysis for new
environmental technologies - the case for biopolymer technology. Journal of
Cleaner Production 17 (2009) 644 - 652.
-G-
G. Bannach, R. Arcaro, D. C. Ferroni, A. B. Siqueira, O.
Treu-Filho, M. Ionashiro, E. Schnitzler. Thermoanalytical study of some
anti-inflammatory analgesic agents. J Therm. Anal. Calorim. 102 (2010)
163-170.
G. Gaucher, M. Poreba, F. Ravenelle, J.-C. Leroux.
Poly(N-vinyl-pyrrolidone)-block-poly( D,L-lactide) as polymeric emulsifier for
the preparation of biodegradable nanoparticles. Journal of Pharmaceutical
Sciences, 96 (7) (2007) 1763-75.
G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger,
J.-C. Leroux. Block copolymer micelles : preparation, characterization and
application in drug delivery. Journal of Controlled Release, 109 (2005)
169-188.
G. Gaucher. Mise au point de nanoparticules polymères pour
l'administration d'agents anticancéreux hydrophobes, Thèse
doctorat, Université de Montréal (2009).
G. J. Atkin, P. Drew, J. L. Turner. Pharmaceutical compsns.
prodn. in the form of homogeneous agglomerates by mixing specified amts. of
2-(4-isobutylphenyl) propionic acid and starch. Patent, WO9304676 (1993).
G. M. Khan, F. Wazir, J.-B. Zhu. Ibuprofen-â-cyclodextrin
inclusion complexes: Evaluation of different complexation methods. The Sciences
1(4) (2001) 193-199.
G. Plucker. Forme pharmaceutique d'ibuprofène. Bruvet
européen n° 0881899. The Boots Company PLC. OFFICE KIRKPATRICK
(2004).
G. S. Kwon. Polymeric drug delivery systems. Taylor & Francis
(ed.). Madison, Wisconsin, USA (2005).
G. W. Cleary. Transdermal controlled-release systems. In Medical
Application of Controlled Release, 1 (1984) 203-251.
-H-
H. A. Garekani, F. Sadeghi, A. Badiee, S. A. Mostafa, A. R.
Rajabi-Siahboomi. Crystal habit modifications of ibuprofen and their
physicomechanical characteristics. Drug Development and Industrial Pharmacy
27(8) (2001) 803-809.
H. Cheng, J. D. Rogers, J. L. Demetriades, S. D. Holland, J.
R. Seibold, E. Depuy. Pharmacokinetics and bioinversion of ibuprofen
enantiomers in humans. Pharmaceutical Research 11 (1994) 824-830.
H. Gelderblom, J. Verweij. Cremophor EL: the drawbacks and
advantages of vehicle selection for drug formulation. Eur J Cancer 37 (2001)
1590-1598.
H. Gollwitzer, K. Ibrahim, H. Meyer, W. Mittelmeier, R. Busch,
A. Stemberger. Antibacterial poly(D,L-lactic acid) coating of medical implants
using a biodegradable drug delivery technology. Journal of Antimicrobial
Chemotherapy 51 (2003) 585-591.
H. Kakula, H. Schlaad, M. Antonietti, S. Förster. The
formation of polymer vesicles or «peptosomes» by
polybutadiene-block-poly(L-glutamate)s in dilute aqueous solution. J. Am. Chem.
Soc. 124(8) (2002) 1658-1663.
H. Laroui. Nanosphères polymères à
couverture de hyaluronate pour la délivrance ciblée de
molécules actives dans le traitement des affections du cartilage,
Thèse doctorat, Université de HENRI POINCARE-NANCY I (2007).
H. N. Rabetafika, M. Paquot, P. Dubois. Les polymères
issus du végétal : matériaux à
propriétés spécifiques pour des applications
ciblées en industrie plastique. Biotechnol. Agron. Soc. Environ. 10(3)
(2006) 185-196.
H. Pranamuda, Y. Tokiwa, H. Tanaka. Polylactide degradation by an
Amycolatopsis sp. Appl. Environ. Microbiol. 63 (1997) 1637-1640.
H. Q. Xie, D. Xie. Molecular design, synthesis and properties of
block and graft copolymers containing polyoxyethylene segments. Prog. Poly.
Sci. 24 (1999) 275-313.
H. Ringsdorf. Structure and properties of pharmacologically
active polymers. J. Polym. Sci. Symp. 51 (1985) 135-153.
-I-
I. Tegeder, J. Pfeilschifter, G. Geisslinger.
Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J., 15
(2001) 2057-72.
-J-
J. A. Richardson, DVM; R. A. Balabuszko, CVT.
Ibuprofen Ingestion in Ferrets: 43 Cases. The Journal of Veterinary Emergency
and Critical Care (2001).
J. Bidone, A. P. P. Melo, G. C. Bazzo, F. Carmignan, M. S.
Soldi, A. T. N. Pires, E. Lemos- Senna. Preparation and characterization of
ibuprofen-loaded microspheres consisting of poly(3-hydroxybutyrate) and methoxy
poly(ethylene glycol)-b-poly(D,L-lactide) blends or poly(3-hydroxybutyrate) and
gelatin composites for controlled drug release. Materials Science and
Engineering C 29 (2009) 588-593.
J. Cho, S. Baratiana, J. Kim, F. Yeh, B. S. Hsiao, J. Runt.
Crystallization and structure formation of poly(L-lactide-co-meso-lactide)
random copolymers: a time-resolved wide- and small-angle X-ray scattering
study. Polymer 44 (2003) 711-717.
J. Folkman, D. M. Long. The use of silicone rubber as a carrier
for prolonged drug therapy, Surg. Res. 4 (1964) 139-142.
J. Jean-François. Synthése et évaluation in
vivo de microparticules d'hydrogel, Thèse doctorat, Université du
QUEBEC à MONTREAL (2004).
J. Khandare, R. Haag. Pharmaceutically used polymers:
principles, structures, and applications of pharmaceutical delivery systems. M.
Schäfer-Korting (ed.), Drug Delivery, Handbook of Experimental
Pharmacology. Springer-Verlag Berlin Heidelberg, 197 (2010) 221-250.
J. L. Grem. 5-Fluoropyrimidines. Cancer chemotherapy &
biotherapy. Principles and practice. B. A. Chabner and D. L. Longo.
Philadelphia, Lippincott Williams &Wilkins (2006) 125-182.
J. Lademann, F. Knorr, H. Richter, U. Blume-Peytavi, A. Vogt,
C. Antoniou. Hair follicles: an efficient storage and penetration pathway for
topically applied substances. Skin Pharmacol Physiol. 21 (2008) 150-155.
J. Namur, M. Wassef, J. P. Pelage, A. Lewis, M. Manfait, A.
Laurent. Infrared microspectroscopy analysis of ibuprofen release from drug
eluting beads in utirine tissue. Journal of Controlled Release 135 (2009)
198-202.
J. Pommay, H. Bouvrais. Formulation, administration et
libération des anti-douleurs. Le MiDiFABs 5 (2006) 59-74.
J. W. Fara. Colonic drug absorption and metabolism, in Novel
Drug Delivery and Its Therapeutic Application, Precott, L. F. and Nimmo, W. S.,
Eds., John Wiley & Sons, Chichester, U. K. (1989).
J.-F. Zhang, X. Sun. Poly(lactic acid)-based bioplastics. Ed.
Woodhead Publishing Limited. Kansas State University, USA (2005).
-K-
K. D. Rainsford. Ibuprofen : A critical bibliographic review.
Taylor & Francis (ed.), London (1999).
K. D. Rainsford. Ibuprofen: pharmacology, efficacy and safety.
Inflammopharmacol 17 (2009) 275-342.
K. J. Jem, J. F. van der Pol, S. de Vos. Microbial lactic
acid, its polymer poly(lactic acid), and their industrial applications. G.-Q.
Chen (ed.), Plastics from bacteria: Natural Functions and Applications,
Microbiology Monographs, 14 (2010).
K. M. Nampoothiri, N. R. Nair, R. P. Jhon. An overview of the
recent developments in polylactide (PLA) research. Bioressource Technology 101
(2010) 8493-8501.
K. Nishimura, S. Nishimura, H. Seo, N. Nishi, S. Tokura, I.
Azuma. Macrophage activation with multiporous beads prepared from partially
deacetylated chitin. Biomed. Mater. Res. 20 (9) (1986) 1359-1372.
K. P. Stock, G. Geisslinger, D. Loew, W. S. Beck, G. L. Bach
and K. Brune. S-Ibuprofen versus ibuprofen-racemate: A randomized double-blind
study in patients with rheumatoid arthritis. Rheumatol. Int. 11 (1999)
199-202.
K. R. Kamath, K. Park. Biodegradable hydrogels in drug delivery.
Drg. Deliv. Rev. 11 (1993) 59-84.
K. W. Kim, S. I. Woo. Synthesis of high-molecular weight
poly(L-lactic acid) by direct polycondensation. Macromol. Chem. Phys. 203
(2002) 2245-2250.
-L-
L. Brannon-Preppas. Biomaterials: Polymers in controlled drug
delivery. Medical Plastics and Biomaterials Magazine (1997).
L. Ilium, P. Watts, A. N. Fisher,
I. labbal-Gill. et Davis S.S.
Novel chitosan based delivery systems for nasal administration of a
LHRH-analogue. STP Pharma LO (2000) 89-94.
L. Xiong, H. W. Jiang, D. Z. Wang. Synthesis, characterization
and degradation of
poly(DLlactide)-block-polyvinylpyrrolidone-block-poly(DL-lactide) copolymers. J
Polym Res 16 (2009) 191-197.
L. Yu. Biodegradable polymer blends and composites from renewable
resources. A John Wiley & Sons, INC, Publication (2009).
L. Y. Qiu, K. J. Zhu. Design of a core-shelled polymer cylinder
for potential programmable drug delivery. International Journal of
Pharmaceutics. 219(1) (2001) 151-160.
L. Y. Qiu, Y. H. Bae. Polymer architecture and drug delivery.
Pharmaceutical Research, 23(1) (2006) 1-30.
L.-T. Lim, R. Auras, M. Robino. Processing technologies for
poly(lactic acid). Progress in Polymer Science 33 (2008) 820-852.
-M-
M. Ajioka, H. Suizu, C. Higuchi, T. Kashima. Aliphatic
polyester and their copolymers synthesized through direct condensation
polymerization. Polym Degrad Stabil; 59 (1998) 137-143.
M. Ajioka, K. Enomoto, K. Suzuke, A. Yamaguchi. Basic
properties of polylactic acid produced by the direct polycondensation
polymerization of lactic acid. Bull Chem Soc Jpn 68 (1995) 2125-2131.
M. Babazadeh. Synthesis and study of controlled release of
ibuprofen from the new acrylic type polymers. International Journal of
Pharmaceutics 316 (2006) 68-73.
M. F. Gonzalez, R. A. Ruseckaite, T. R. Cuadrado. Structural
changes of polylactic-acid (PLA) microspheres under hydrolytic degradation.
Journal of Applied Polymer Science, (71) (1999) 1223-1230.
M. J. Vincent, H. Ringsdorf, R. Duncan. Polymer therapeutics :
Clinical applications and challenges for development. Advanced Drug Delivery
Reviews 61 (2009) 1117-1120.
M. M. Arons, A. P. Wheeler, G. R. Bernard, B. W. Christman, J.
A. Russell, R. Schein, W. R. Summer, K. P. Steinberg, W. Fulkerson, P. Wright,
W. D. Dupont, B. B. Swindell. Effects of ibuprofen on the physiology and
survival of hypothermic sepsis. Ibuprofen in Sepsis Study Group. Crit Care
Med., 27(4) (1999) 699-707.
M. Manish, J. Harshal, P. Anant. Melt sonocrystallization of
ibuprofen: Effect on crystal properties. European Journal of Pharmaceutical
Sciences 25 (2005) 41-48.
M. Melillo, G.J. Phillips, J.G. Davies, A.W. Lloyd, S.R.
Tennison, O.P. Kozynchenko, S.V. Mikhalovsky. The effect of protein binding on
ibuprofen adsorption to activated carbons. Carbon 42 (2004) 565-571.
M. Moneghini, B. Bellich, P. Baxa, F. Princivalle. Microwave
generated solid dispersions containing Ibuprofen. International Journal of
Pharmaceutics 361 (2008) 125-130.
M. Mort. Multiple modes of drug delivery. Modern drug discovery.
3(3) (2000) 30-32.
M. Schäfer-Korting, W. Mehnert, H. C. Korting. Lipid
nanoparticles for improved topical application of drugs for skin diseases. Adv
Drug Deliv Rev. 59 (2007) 427-443.
M. Shimada, S. Natsugoe, T. Kumanohoso, T. Aikou, H. Shimazu,
K. Nakamura. Local chemotherapy of esophageal cancer with bleomycin adsorbed to
activated carbon particles. In: Nabeya K, Hanaoka T, Nogami H (eds) Recent
advances in diseases of the esophagus. Springer, Tokyo, (1993) 905.
M. T. Razzak, D. Darwis, Zainuddin and Sukimo. Irradiation of
polyvinyl alcohol and polyvinyl pyrrollidone blended hydrogel for wound
dressing. Radiat. Phys. Chem. 62 (2001) 107-113.
M.-C. Jones, M. Ranger, J.-C. Leroux. pH-sensitive unimolecular
polymeric micelles: synthesis of a novel drug carrier. Bioconjug. Chem. 14
(2003) 774-781.
M. Vert, I. Dos Santos, S. Ponsart, N. Alauzet, J.-L. Morgat,
J. Coudane, H. Garreau. Degradable polymers in living environment: Where do you
end up. Polymer International 51 (2002) 840-844.
-N-
N. A. Peppasa, P. Buresa, W. Leobandunga, H. Ichikawab.
Hydrogels in pharmaceutical formulations. European Journal of Pharmaceutics and
Biopharmaceutics 50 (2000) 27-46. N. B. Graham. Polyethylene glycol chemistry.
In: Harris JM., editor. Biotechnical and Biomedical Application. New York:
plenum Press; (1992) 1-13.
N. E. Suyatma, Developpement de films biodegradables à
base de chitosane: Etudes du Mélange Chitosane/PLA, de la Plastification
et de la Compatibilisation, Thèse doctorat, Universite de Reims -
Champagne ardenne (2006).
N. Hasirci. Micro and nanosystems in biomedicine and drug
delivery. M. R. Mazafari (ed.), Nanomaterials and Nanosystems for Biomaterials
Applications (2007) 1-26.
N. Lucas, C. Bienaime, C. Belloy, M. Queneudec, F. Silvestre,
J.-E. Nava-Saucedo. Polymer biodegradation : Mechanisms and estimation
techniques. Chemosphere 73 (2008) 429-442.
N. M. Davies. Clinical Pharmacokinetics of Ibuprofen. The first
30 years. Clin Pharmacokinet, 34 (1998) 101-54.
N. V. Phadnis, R. Suryanarayanan. Simultaneaous quantification
of an enantiomer and racemic compound of ibuprofen by X-ray powder
diffractometry. Pharmaceutical Research 14(9) (1997).
-O-
O. Hung. Drug transformation : Advances in drug delivery systems.
CAN. J. ANESTH. 53 (11) (2006) 1074-1077.
-R-
R. A. Grinsted, J. L. C. Koenig. Study of cyclic
sorption-desorption into poly(methylmethacrylate) rods using RMN imaging.
Macromolecules. 25 (1992) 1235-1241.
R. Arshady. Microspheres, microcapsules and liposome, Vol. I.
Preparation and Chemical Applications. Citrus, London (1999).
R. Gurny, N. A. Peppas, O. O. Harrington, G. S. Banker.
Development of biodegradable and injectable lattices for controlled delivery of
potent drugs. Drug Dev. Ind. Pharm. 7 (1981) 1- 25.
R. K. Yeh, J. Chen, J. L. Williams, M. Baluch, T. R. Hundley,
R. E. Rosenbaum, S. Kalala, F. Traganos, F. Benardini, P. del Soldato, K.
Kashfi, B. Rigas. NO-donating nonsteroidal antiinflammatory drugs (NSAIDs)
inhibit colon cancer cell growth more potently than traditional NSAIDs: a
general pharmacological property. Biochem Pharmacol 67 (2004) 2197-2205.
R. Kircheis, L. Wightman, M. Kursa, E. Osterman, E. Wagner.
Tumor-targeted gene delivery : an attractive strategy use highly active effect
for molecules in cancer treatment, Gene Ther., 9 (2002) 731-735.
R. Langer, J. Folkman. Polymers for the sustained release of
proteins and other macromolecules. Nature, 263 (1976) 797-800.
R. M. Rasal, A. V. Janorkar, D. E. Hirt. Poly(lactic acid)
modifications. Process in Polymer Science 35 (2010) 338-356.
R. Pignatello, M. Ferro, G. Puglisi. Preparation of Solid
Dispersions of Nonsteroidal Antiinflammatory Drugs With Acrylic
Polymers and Studies on Mechanisms of Drug-Polymer Interactions. AAPS
PharmSciTech 3 (2) (2002).
R. S. Blackburn, DW Farrington, J Lunt, S Davies. Biodegradable
and sustainable fibers. University of Leeds, United Kingdom, (2005).
-S-
S. Bogdanova, I. Pjeva, P. Nikolova, I. Tsakovska, B.
Müller. Interactions of poly(vinylpyrrolidone) with ibuprofen and
naproxen: Experimental and modeling studies. Pharmaceutical Research 22(5)
(2005) 806-815.
S. Davaran, A. A. Entezami. Synthesis and Hydrolysis of Modified
Polyvinyl Alcohols Containing Ibuprofen Pendant Groups. Iranian Polymer Journal
5(3) (1996).
S. Freiberg, X. X. Zhu. Polymer microspheres for controlled drug
release. International Journal of Pharmaceutics 282 (2004) 1-18.
S. G. Kazarian, G. G. Martirosyan. Spectroscopy of
polymer/drug formulations processed with supercritical fluids: in situ ATR-IR
an Raman study of impregnation of ibuprofen into PVP. International Journal of
Pharmaceutics 232 (2002) 81-90.
S. J. De Jong, E. R. Arias, D. T. S. Rijkers, C. F. Van
Nostrum, J. J. Kettenes-Van Den Bosch, W. E. Hennink. New insights into the
hydrolytic degradation of poly(lactic acid): participation of the alcohol
terminus. Polymer 42 (2001) 2795-2802.
S. Lakshmi, C. T. Laurencin. Polymers as biomaterials for tissue
engineering and controlled drug delivery. Adv. Biochem. Engin/Biotechnol. 102
(2006) 47-90.
S. Lerdkanchanaporn, D. Dollimore. A thermal analysis study of
ibuprofen. Journal of Thermal Analysis, 49 (1997) 879-886.
S. Madival, R. Auras, S. P. Singh, R. Narayan. Assessment of
the environmental profile of PLA, PET and PS clamshell containers using LCA
methodology. Journal of Cleaner Production 17 (2009) 1183-1194.
S. Nakayama, K. Ihara, M. Senna. Structure and properties of
ibuprofen-hydroxypropyl methylcellulose nanocomposite gel. Powder Technology
190 (2009) 221-224.
S. Shukla, A. K. Bajpai, J. Bajpai. Swelling controlled
delivery of antibiotic from a hydrophilic macromolecular matrix with
hydrophobic moieties. Macromol Res. 11(4) (2003) 273-282.
-T-
T. Allen, P. Cillis. Drug delivery systems : entering the
mainstream, Science, 303 (2004) 1818-1822.
T. Hayashi, Y. Ikada. Protease immobilization onto porous
chitosan beads. 1. Appl. Polym. Sei. 42 (1991) 85-92.
T. Higuchi. Design of chemical structure for optimal dermal
delivery. Curr. Prob. Dermatol., 7 (1978) 121.
T. J. Kreeger. Overview of delivery systems for the
administration of contraceptives to wildlife. University of Nebraska-Lincoln
(1993).
T. Kumanohoso, S. Natsugoe, M. Shimada, T. Aikou. Enhancement
of therapeutic efficacy of bleomycin by incorporation into biodegradable
poly-d,l-lactic acid Cancer Chemother Pharmacol 40 (1997) 112-116.
T. Lammers, W. E. Hennink. Tumour-targeted nanomedicines:
principles and practice. Br J Cancer 99(3) (2008) 392-7.
T. Niwa, H. Takeuchi. Preparations of biodegradable
nanospheres of water soluble and insoluble drugs with D,L-lactide/glycolide
copolymer by a novel spontaneous emulsification solvent diffusion method, and
drug release behavior. J. Controlled Release 25 (1993) 89-98.
T. Ohkhita, S. H. Lee. Thermal degradation and biodegradability
of poly(lactic acid)/corn starch biocomposites. J. Appl. Polym. Sci. 100 (2006)
3009-3017.
T. Phromsopha, Y. Baimark. Methoxy poly(ethylene
glycol)-b-poly(D,L-lactide) films for controlled release of ibuprofen. Trends
in Applied Sciences Research 4(2) (2009) 107-115.
T. Vasconcelos, B. Sarmento, P. Costa. Solid dispersions as
strategy to improve oral bioavailability of poor water soluble drugs. Drugs
Discovery Today. 12 (2007) 1068-1075.
T. Yamaoka, Y. Takahashi, T. Ohta, M. Miyamoto, A. Murakami,
Y. Kimura. Synthésis and proprties of multiblock copolymers consisting
of poly(L-lactic acid) and poly(oxypropyleneco-oxyethylene) prepared by direct
polycondensation. Journal of Polymer Science : Part A : Polymer Chemistry, Vol.
37, 1513-1521 (1999).
T. Zecheru. New biopolymers with possible use in the field of
dentistry and in the field of orthopaedics, Thesis doctorat, University
Polithdcnica of Bucharest, ROMANIA (2008).
-V-
V. Michel, M. Jacques, B. Niels. Implant biorésorbable
à base d'acide polylactique contenant un antibiotique. Fascicule de
Brevet Européen. Centre National de la Recherche Scientifique (CNRS),
France (1993).
V. P. Torchilin. Structure and design of polymeric
surfactant-based drug delivery systems. J. Control. Release 73 (2001)
137-172.
V. P. Torchilin. Targeted polymeric micelles for delivery of
poorly soluble drugs. CMLS, Cel. Mal. Life Sci. 61 (2004) 2549-2559.
V. T. R. Ho, R. G. Blank. Therapeutic taste-neutral powder
form of Ibuprofen obtd. By spraying-drying dispersion of Ibuprofen and ethyl
cellulose in water contg. dissolved or suspended. Patent, EP-322137 (1989).
V. V. Ranade, M. A. Hollinger. Drug delivery systems.
2nd edition, by CRC Press LLC (2004).
V. V. Ranade a. Drug delivery systems, 5B, Orale drug
delivery, J. Clin. Pharmacol., 31 (98) (1991).
V. V. Ranade b. Drug delivery systems. 5A. Orale drug
delivery, J. Clin. Pharmacol., 31 (98) (1991).
-W-
W. Holubek, A. Stolbach, S. Nurok, O. Lopez, A. Wetter, L.
Nelson. A Report of Two Deaths from Massive Ibuprofen Ingestion. Journal Of
Medical Toxicology. 3(2) (2007).
W. Hoogsteen, A. R. Postema, A. J. Pennings, G. ten Brinke, P.
Zugenmaier. Crystal Structure, Conformation, and Morphology of Solution-Spun
Poly(L-lactide) Fibers. Macromolecules 23 (1990) 634-642.
-X-
X. Kaitian, A. Kozloca, E. B. Denkbas, E. Piskin. Poly(D,L-lactic
acid) homopolymers : Synthesis and characterization. Tr. J. of Chemistry 20
(1996) 43-53.
X. Shuai, T. Merdan, A. K. Shaper, F. Xi, T. Kissel.
Core-Cross-linked polymeric micelles as paclitaxel carriers. Bioconjug. Chem.
15 (2004) 441-448.
-Y-
Y. Cheng, S. Deng, P. Chen, R. Ruan. Polylactic acid synthesis
and modifications : a review. Front. Chem. China, 4(3) (2009) 259-264.
Y. J. Manrique, D. P. Pacheco, F. Martinez. Thermodynamics of
mixing and solvation of ibuprofen and naproxen in propylene glycol-water
cosolvent mixtures. J. Solution Chem 37 (2008) 165-181.
Y. Oda, A. Yonetsu, T. Urakami, K. Tomomura. Degradation of
polylactide by commercial proteases. J. Polym. Environ. 8 (2000) 29-32.
Y. Tokiwa . B. P. Calabia. Biodegradability and biodegradation of
poly(lactide). Appl Microbiol Biotechnol 72 (2006) 244-251.
Y. X. Li, J. Nothnagel, T. Kissel. Biodegradable brush like
graft polymers from poly(D,Llactide) or poly(D,L-lactide-co-glycolide) and
charge-modified, hydrophilic dexrans as backbone synthesis, characterization
and in vitro degradation properties. Polymer 38 (25) (1997) 6197-6206.
-Z-
Z. Zhong-cheng, Ruan Jian-ming, Huang Bai-yun, Li Ya-jun, Zou
Jian-peng, Zhang Hai-bo. Preparation and characterization of poly(D,L-lactide)
and its porous biomaterials. J. Cent. South Univ. Technol. 12 (1) (2005)
1-4.
Résumé
Résumé :
Ce travail se place dans le contexte de la synthèse
d'un polymère biodégradable issu de ressources renouvelables
comme le maïs, et son application dans le domaine biomédicale, en
particulier comme vecteur de principes actifs. Trois polymères de type
poly(DL-acide lactique) (PDLLA) de différents poids moléculaires
(1000, 3000 et 9000) g/mol ont été synthétisés par
la méthode de polymérisation par polycondensation
azéotropique de D,L-acide lactique à 140°C sous vide en
présence d'un catalyseur (SnCl2). Les masses moléculaires des
différents polymères ont été
déterminées par la méthode viscosimétrique, par
l'utilisation d'un viscosimètre capillaire (Ubbelhode).
Par la suite, des mélanges PDLLA/ibuprofène (IB)
ont été préparés par l'utilisation de trois
méthodes de préparations qui sont méthode physique, fusion
à chaud et par évaporation du solvant. Pour chaque type de
mélange, trois formulations (F5, F15 et F25) ont été
préparées. Toutes les formulations contiennent la même
quantité d'IB (20 mg) mais elles diffèrent de la quantité
de PDLLA incorporée 5% (F5), 15% (F15) et 25% (F25).
Les produits bruts (PDLLA, IB) ainsi que les
différentes formulations ont été
caractérisés en utilisant plusieurs méthodes de
caractérisations physiques telles IRTF, DRX, MEB et DSC. Nous avons
constaté que, une fois les particules de l'IB sont
mélangées avec celles de PDLLA, la taille et la morphologie des
particules ont été altérées. En plus, la
méthode IRTF nous a révélé que la bande
d'absorption dans la région des carbonyles de l'ibuprofène a
été décalée vers des fréquences (í)
supérieures, une fois l'IB est mélangé avec le PDLLA. En
effet, des interactions par ponts hydrogènes ont été
supposées existantes entre l'IB et PDLLA. La diffraction des rayons X
nous a révélé que le spectre de l'ibuprofène a subi
certains changements la largeur, la hauteur et la position des pics.
Les résultats de dissolution in vitro ont montré
que la vitesse de libération de l'ibuprofène a été
modifiée dans toutes les formulations. L'ibuprofène a
été libéré d'une manière rapide au
début, suivi par une libération lente (ou profil quick/slow). La
vitesse de libération de l'IB diminue quand la quantité et la
masse moléculaire de PDLLA augmentes. La cinétique de
libération de l'IB est plus rapide dans un milieu de dissolution alcalin
(pH 7,4) que dans un milieu acide (pH 5,8). La cinétique de
libération de l'ibuprofène ralentisse aussi en fonction de la
méthode de préparation dans l'ordre : mélange par fusion
à chaud, mélange physique et mélange par
évaporation du solvant.
Mots clés :
Poly(D,L-acide lactique) (PDLLA), Ibuprofène, Polymère
biodégradable, Vecteur de principes actifs, Mélange
physique, Mélange par
fusion à chaud, Mélange par évaporation du
solvant.
Abstract
Abstract :
This work it puts in the context of the synthesis of a
biodegradable polymer derived from renewable resources such as corn, and its
application in biomedical area, particularly as drug carriers. Three
poly(D,L-lactic acid) polymers with different molecular weights (1000, 3000 and
9000) g/mol were synthesized by a method of azeotropic condensation
polymerization of D,L-lactic acid at 140°C under vacuum with using a
catalyst (SnCl2). Molecular weights of PDLLAs were determined by a viscosimeter
(Ubbelhode).
Thereafter, PDLLA/ibuprofen (IB) mixtures were prepared using
three methods of preparation such as physical mixes, hot mixes and solvent
evaporation methods. For each mixture, three formulations (F5, F15 et F25) were
prepared. All the formulations contain the same quantity of IB (20 mg) but they
differ from the quantity of PDLLA loaded 5% (F5), 15% (F15) et 25% (F25).
The crude compounds (PDLLA, IB) as well as the different
formulation were characterized by using several characterization physical
methods as FTIR, XRD, SEM and DSC. We noted that, once the particles of IB are
mixed with those of PDLLA, size and morphology of particles were altered.
Moreover, FTIR methods revealed that the absorption band in the region of
carbonyl of ibuprofen was shifted towards the higher frequencies (í),
once IB is mixed with PDLLA. Indeed, interactions with hydrogen with hydrogen
bonding were supposed to be existing between IB and PDLLA. The X-ray
diffraction revealed that the spectrum of ibuprofen undergo certain changes
such as widening of the width, increase height, and shift of the peaks.
The results of in vitro dissolution showed that the rate
release of ibuprofen was modified in all the formulations. Ibuprofen was
released in fast way at the first stage, followed by slow release (or
quick/slow profile). The rate release of the IB decreases when the quantity and
the molecular weights of PDLLA increases. The release kinetics of IB is faster
in an alkaline medium of dissolution (pH 7.4) that in an acidic one (pH 5.8).
The release kinetics of ibuprofen also slows down according to the method of
preparation in the order : hot mix, physical mix and solvent evaporation
mix.
Keywords: Poly(D,L-lactic acid)
(PDLLA), Ibuprofen, Biodegradable polymers, drug carriers, physical mixes, hot
mixes, solvent evaporation methods.
| 

