2.1.3. Différentes parties du cancer du colon
2.1.3.1. Cancers du colon droit
Ils représentent 40% des cancers de colon, on les
observe essentiellement chez l'homme de 50 à 60 ans et ils se rangent
parmi les tumeurs malignes. Il s'agit d'épithéliomas glandulaires
typiques du moins en générale, l'infection est fréquente.
Ils siégent de la valvule iléo-caecale au tiers droit du
transverse (Puig, 1997).
Ce cancer, qui intéresse la portion du cadre colique
dont le calibre est le plus large, est parmi les plus latents, avec un faible
risque d'occlusion, et une infection fréquente. Les tumeurs basses
caecales sont le plus souvent bourgeonnantes, alors que les tumeurs proches de
l'angle droit sont plus volontiers rétractiles et squirreuses
(Bonithon, 1999).
2.1.3.2. Cancers du colon transverse
Si leur description macroscopique est comparable à
celle des segments coliques adjacents, leur propagation lymphatique est
particulière et de mauvais pronostic. L'envahissement ganglionnaire peut
se faire vers l'origine des deux axes vasculaires mésentérique
supérieur et mésentérique inférieur, et atteint
précocement les ganglions rétro pancréatiques,
inaccessibles à une exérèse chirurgicale curative
(Puig, 1997).
Le cancer du colon transverse se caractérise par le
fréquence de la symptomatologie d'emprunt souvent trompeuse, crises
coliques, troubles du transit, distension caecale, ballonnements, palpations
d'une tumeur épigastrique, ou tout
autre signe pouvant orienter vers une affection gastrique,
biliaire ou pancréatique (Bonithon, 1999).
2.1.3.3. Cancers du colon gauche
Ce sont les plus fréquents des cancers coliques, dont ils
représentent 60% de l'ensemble, et les 3/4 d'entre eux siègent
sur le sigmoïde (Puig, 1997).
Ils sont rapidement sténosants avec symptomatologie
occlusive. Très lymphophiles, ils essaiment vers l'origine de
l'artère mésentérique inférieure qui doit
être liée à sa naissance devant l'aorte, lorsque
exérèse chirurgicale se veut curative, permettant un curage
ganglionnaire complet de l'axe mésentérique inférieur. Le
cancer de l'angle gauche se caractérise par la précocité
et la fréquence de la propagation lymphatique rétro
pancréatique (Bonithon, 1999).
2.1.4. Classification et stadification
De nombreuses classifications à visée pronostic
ont été proposées pour les cancers colorectaux.
o Classification de Dukes (1967)
A : envahissement de la sous muqueuse
B : envahissement de musculaire sans envahissement
ganglionnaire
C : métastase ganglionnaire
D : métastase à distance
o Classification d'Astler Coller (1954) A : ne
dépasse pas la sous muqueuse
B1 : envahissement de la musculaire N-
B2 : atteinte de la séreuse
C1 : ne dépasse pas la séreuse, envahissement
ganglionnaire N+
: dépasse la séreuse, envahissement ganglionnaire
N+
o Classification TNM
La classification TNM est un système international,
proposé par le chirurgien français Pierre Denoix dans les
années 1940B1950, de façon à classer les cancers selon
leur extension anatomique (Denoix, 1946).
Les trois lettres symbolisent la propagation de la maladie
cancéreuse sur le site de la tumeur primitive (T), dans les ganglions
lymphatiques voisins (N pour node en anglais) et à distance pour
d'éventuelles métastases (M). Chaque lettre est affectée
d'un coefficient. Dans son principe, cette classification considère
seulement les données cliniques et ne s'applique qu'à des cancers
qui n'ont pas encore été traités. (Fig
11)
T (tumeurs)
Tis intra épithéliale ou chorion
T1 sous muqueuse
T2 musculeuse
T3 à travers les musculaires propria dans la sous
séreuse ou dans les tissus péri coliques non
péritonéales.
T4 organe ou structure de voisinage et/ou perforation du
péritoine viscérale.
N (ganglions)
N0 pas de métastase ganglionnaire
Nx ganglions non évalués ou moins de 8 ganglions
examinés N1 à N3 ganglions métastasiques
régionaux
N2, N4 ganglions métastasiques régionaux ou plus.
M (métastases)
M0 pas de métastase
M1 métastases à distances
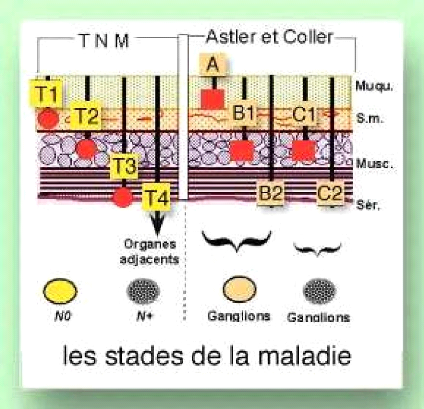
Figure 11: comparaison entre les classifications TNM
et
Astler Coller.
o Stadification
A partir des données de classification TNM, les
cancers du colon sont classés en quatre stades. Les chances de
guérison varient considérablement du stade I au stade IV
(tableau II) (Oconnell et a!, 2004).
Pour chacun des stades est noté le taux de survie
après le traitement.
Tableau II : stadification des cancers
colorectaux.
(Oconnell et a!, 2004).
Stade
|
Classification TNM
|
Taux de survie
|
I
|
T1-T2-N0-M0
|
93.2%
|
II A
|
T3-N0-M0
|
84.7%
|
II B
|
T4-N0-M0
|
72.2%
|
III A
|
T1-T2-N1-M0
|
83.4%
|
III B
|
T3-T4-N1-M0
|
64.1%
|
III C
|
Tous T N2-M0
|
44.3%
|
IV
|
Métastases à distance
|
8.1%
|
|
1. Apport du dosage de l'antigène carcinome
embryon dans le cancer du colon
Le nom antigène carcino-embryonnaire (ACE) a
été donné en 1965 par Gold et Freedman à une
protéine normalement exprimée par le tube digestif du foetus
durant les six premiers mois de gestation, et retrouvée dans les cancers
du pancréas, du foie et du colon (Gold, 1965).
1.1. Description
1.1.1. Structure
L'ACE est une glycoprotéine de poids
moléculaire compris entre 180 et 200 KDa, fortement glycosylée.
Vingt huit sites de N-glycosylation sur des résidus aspargine sont
présents dans le domaine extracellulaire de la molécule.
L'ACE appartient à la super famille des
immunoglobulines dont plusieurs membres sont impliqués dans le processus
d'adhérence et de reconnaissance intercellulaire.
L'ACE possède un domaine N-terminal de 108 acides
aminés, similaire aux domaines variables des immunoglobulines et des
domaines An et Bn analogues aux domaines des Ig (Hammarstrom,
1999).
1.1.2. Gènes
Le gène de l'ACE est le CEA-related cell adhesion
molecule 5 (CEACAM) (Beauchemin, 1999). L'ACE est le chef de file d'une famille
de glycoprotéines qui sont issues, chez l'homme et chez les primates, de
la transcription de 29 gènes localisés sur le bras long du
chromosome 19 dans la région 19q13.1-19q13.3.
L'analyse nucléotidique permet de diviser des
gènes en trois sous groupes. Le sous groupe de l'ACE qui comprend 12
membres, le sous groupes Pregnancy specific glycoprotéins (PSG) qui
comprend 11 membres et un troisième sous groupe qui comprend six pseudos
gènes.
L'expression des gènes du sous groupe ACE donne des
protéines ancrées à la membrane par un lien phosphatidyl
inositol (dont la molécule ACE et le non-specific
Gross-reacting antigen « NCA ») et des
protéines transmembranaires possédant un domaine cytoplasmique
(dont les biliary glycoprtein) (Hammarstrom, 1999).
1.1.3. Synthèse
Chez l'adulte, l'ACE est synthétisé
principalement dans certaines portions du tube digestif tel que la langue,
l'oesophage distal, l'estomac, l'intestin grêle. Le colon et le rectum
où il n'est présent qu'au pole apical des cellules
épithéliales.
Chez le foetus, la synthèse est abondante au niveau du
duodénum et du colon à partir de la 9éme semaine, à
un stade où l'épithélium n'est pas encore
stratifié, l'antigène est alors localisé sur toute la
surface de la membrane cellulaire, la stratification de
l'épithélium coïncidant avec la restriction de l'ACE au
domaine apical de la cellule (Létourneau, 1997).
Dans le cancer du colon, le gène ACE est sur
exprimé. Le gradient de sécrétion vers le pole apical est
perturbé et l'antigène carcino-embryonnaire est distribué
sur toute le surface de la cellule et dans l'espace intercellulaire
(Benchimol, 1989) ;(Jothy, 1993).
In vitro, l'expression de l'ACE peut être
augmentée dans certaines lignées de tumeurs coliques par les
cytokines telles que l'interféron gamma (Guadagin,
1990), les « transforming growth factor » béta1 et
béta2 (Chakrabarty, 1988) ;(Chakrabarty,
1990), l'interleukine6 (Ullmann, 1992) ou des
combinaisons, interféron, gammainterféron, alpha-interleukine 6
(Verhaar, 1999). Une régulation par les cytokines a
été également observée in vivo, un traitement par
interféron gamma entraine l'augmentation de la concentration
sérique de l'ACE chez environ 65% des patients traités
(Greiner, 1991).
1.1.4. Récepteurs
Un récepteur spécifique de l'ACE de 80KDa a
été décrit au niveau des cellules de Kupffer du foie
(gangopadhyay.1996). Dans les lignées cellulaires du
cancer du colon ; les galectines ont un rôle dans les interactions
cellulaires, l'adhésion à la matrice extracellulaire et
l'acquisition de phénotype métastatique.
1.2. Fonctions cellulaires
L'ACE est une molécule dotée de
propriétés adhésives, elle est capable d'adhésion
homotypique (ACE-ACE) ou hétéro typiques (ACE-BGP), (ACE, NCA)
avec d'autres membres de la famille des antigènes carcino-embryonnaires,
avec le collagène de type I, ainsi qu'avec certaines souches d'E. Coli.
(Thompson, 1991).
Les fonctions physiologiques de l'ACE sont encore à
préciser. Il jouerait un
rôle
o Dans la différenciation cellulaire au cours de
l'embryogénèse.
o Dans la reconnaissance et la régulation de la flore
bactérienne du colon colon normal (Létourneau,
1997).
La participation active de l'ACE dans la
cancérogénèse et la dissémination
métastatique est actuellement clairement établie. Le
dérèglement de l'expression de plusieurs membres de la famille
des antigènes carcinoembryonnaires est un évènement
précoce dans la cancérogénèse colique.
(Johnson, 1991) ; (Nollau, 1997) ;
(Thompson, 1997) :
o La BGP et le carcinoembryonic Antigen Familly Member2
(CGM2) qui jouerait un rôle de suppresseur de tumeur sont
réprimés. (Neumaier, 1993).
o L'ACE, les NCA 50/90, sont surexprimés, les NCA
suivent le même profil d'expression que l'ACE, avec une fréquence
et une intensité de surexpression qui sont supérieurs à
celles de l'ACE (CHU, 1991).
Il existe une relation entre la production d'ACE par des
lignées cellulaires de cancer colique humain et leur potentiel
tumorigène et métastatique testé par injection intra
splénique de cellule chez la souris nude (Tibbetts,
1993).
Le blocage des molécules d'ACE à l'aide d'un
fragment Fab anti ACE diminue l'incidence des métastases
(Hashino, 1994). La transfection par l'acide
désoxyribonucléique complémentaire (ADNc) d'ACE induit un
phénotype métastatique dans les cellules de mélanomes
(Grimm, 1995).
Différentes expériences conduisent à
formuler des hypothèses sur le rôle joué par l'ACE
présent à la surface des cellules tumorales dans le processus
d'induction des métastases. Des cellules sont non
sécrétrices d'ACE acquièrent la propriété de
s'agréger entre elles. In vitro, l'ACE fonctionne comme une
molécule d'adhésion intercellulaire (Benchimol, 1989).
Il existe une corrélation entre le niveau de
production de l'ACE par les cellules et la quantité
d'agrégats formés. In vivo, les grappes cellulaires pourraient,
en se bloquant dans la microcirculation, favoriser la survenue de
métastases (Hashino, 1994) ; (Thomas, 1995).
In vitro, la fixation de l'ACE sur le
récepteur présent à la surface des cellules de Kupffer
induit la sécrétion des interleukines alpha I, béta1 et 6
qui activent la production de molécules d'adhésion par les
cellules endothéliales (Gangopadhyay, 1996 A). Par ce
mécanisme, l'ACE pourrait faciliter la rétention des cellules
tumorales au niveau du foie (Gangopadhyay. 1996).
Un modèle expérimental reproduisant l'invasion
tumorale montre que l'ACE est capable d'exercer une attraction chimique sur les
cellules de cancer colorectal. En outre, l'ACE faciliterait l'adhésion
des cellules tumorales aux protéines de la matrice
extracellulaire (Kim, 1999).
Bien qu'il ait été montré chez la souris
nude que l'apparition de lésions secondaires au niveau du foie
après injection intra splénique de cellules de cancer colorectal
était facilitée par une administration intraveineuse
préalable d'ACE (Hostetter, 1990), des
expériences récentes concluent que l'ACE libéré
dans la circulation ne participe pas au développement des
métastases hépatiques (Lecante, 1999).
L'ACE peut interférer avec l'immunité
cellulaire anti tumorale dépendante des lymphokines -activated killer
(LAK). Des expériences in vitro montrent que l'expression par les
cellules tumorales des molécules d'ACE les protège de la lyse par
les cellules LAK (Kammerer, 1994). Cette protection passerait
par une interaction avec une molécule présente à la
surface des lymphocytes (Kammerer, 1996). L'hyper expression
de l'ACE et du NCA est en partie responsable de la résistance aux
chimiothérapies. La transfection par l'ADNc d'ACE diminue la
sensibilité des cellules à l'adriamycine (Kawaharata,
1997).
Dans les cellules MCF-7 le phénotype multidrug
résistant sans hyper expression de la P-glycoprotéine ou de la
MPP (multidrug resistance protéin) a pu être rattaché
à la protéine P95 qui a été identifiée comme
étant la NCA. (Ross, 1997).
1.3. Techniques de dosages
Les méthodes initiales de mesure de la concentration
sérique de l'ACE nécessitaient une extraction de
l'antigène préalable à son dosage, ce qui rendait la
détermination relativement peu précise. D'autre part, les
premiers dosages immunologiques étaient basée sur une
reconnaissance avec des anticorps polyclonnaux à l'origine de nombreuses
réactions croisées avec les membres de la famille ACE, en
particulier les NCA. Avant les années 80, les différences de
résultats obtenus chez un même patient avec deux types de
techniques pouvaient être considérables (Fletcher,
1986).
Actuellement de nombreux anticorps monoclonaux capables de ne
pas reconnaître les NCA permettent le dosage de l'ACE sérique par
immunométrie (Hammarstrom, 1989).
La présence dans les échantillons sanguins
humains d'anticorps anti immunoglobulines (facteurs rhumatoïdes....etc.)
est susceptible de produire des interférences sur le dosage, responsable
d'une diminution ou d'une augmentation artificielle des concentrations. De
multiples améliorations techniques ont té apportées par
les fabricants de réactifs afin de minimiser les perturbations dues aux
anticorps. Il importe néanmoins que le biologiste ait conscience de la
présence possible de ces anticorps dans le sang du patient et que le
clinicien avertisse le laboratoire de l'existence de situations favorisants
leur survenue (pathologie auto-immune, injection d'anticorps à des fins
thérapeutiques...etc.) (Grassi, 1994).
1.4. Demi -vie
Le foie est le site principal de la clairance de l'ACE
(anon, 1996). Mesurées chez 295 patients
opérés pour un cancer colorectal, les demi-vies ont une
distribution autour d'une moyenne géométrique de 2,8 intervalle
de confiance 95%, valeurs extrêmes 0,8 à 10 jours. (Carl,
1993). Ces résultats sont en accord avec ceux obtenus pour
d'autres localisations tumorales sécrétant l'ACE.
(Verhelset, 1991) ; (Rapellino, 1994) ;
(Yohinasu, 1997).
1.5. Les valeurs de référence
De la revue générale réalisée par
Fletcher en 1986, il ressort que :
o Les valeurs de référence les plus souvent
admises sont 2,5 et 5tg/l, 84 à 87% des personnes normales ont une
concentration sérique d'ACE 2,5tg/l et 95 à 98 % ont une
concentration sérique d'ACE 5tg/l.
o L'ACE est en moyenne un peu plus élevé chez les
hommes et chez les sujets âgés (Fletcher,
1986).
o Chez les fumeurs l'ACE est 1,6 à 1,7 fois plus
élevé que chez les non
fumeurs (Shahangian, 1991) ; (Verdi, 1993) ; (Patel,
1995).
1.6. Place du dosage de l'ACE dans la prise en charge
des patients atteints du cancer du colon
1.6.1. Dépistage et diagnostic précoce des
cancers colorectaux
La recherche bibliographique a permis d'identifier neuf
études ayant évalué l'intérêt du dosage de
l'ACE pour le dépistage et le diagnostic précoce des cancers
colorectaux en termes de sensibilité et de spécificité.
Ces neufs études retrouvent des valeurs extrêmes pour la
sensibilité de 11 à 40% pour les tumeurs de stades A et B de
Dukes. La spécificité est évaluée entre 70 et 97%
vis-à-vis des maladies bénignes digestives et non digestives
(TAB III), (Carpelamholmstr, 1996, B).
La valeur diagnostique de l'ACE vis-à-vis des cancers
colorectaux est faible en raison de son manque de sensibilité pour les
stades localisés et de la fréquence des concentrations
sériques supérieurs à la valeur de référence
lors des maladies bénignes et des autres localisations
cancéreuses. En outre, des concentrations sériques normales sont
fréquemment associées aux cancers peu différenciés
(Goslin, 1981) ; (Quentmeier, 1987).
L'ASCO et l'ANAES ne recommandent pas de doser l'ACE dans du
dépistage et du diagnostique précoce des cancers colorectaux
(Anon, 1996) ; (Agence nationale.1997).
Même dans des situations de forte prévalence le
dosage de l'ACE ne doit pas être prescrit dans un but de diagnostique
(Fletcher, 1986) ; (Durand, 1990) (tab II).
Tableau III : études ayant évalué
la corrélation des concentrations sériques de
l'ACE avec les
stades d'extension des cancers du colon. (Carpelamholmstr, 1996, B)
Références
|
Marqueur
|
Seuil
|
Sensibilité
% (nombre de patients étudiés)
|
Spécificité %
(nombre de sujets étudiés)
|
|
Sujets sains
|
Maladies bénignes
|
|
Dukes* C et D
|
|
[KUUSELA1984]
|
ACE Tg/L
|
2,5
|
31% (26)
|
82% (45)
|
|
70% (37)
|
[GUPTA1985]
|
|
11% (151)
|
55% (130)
|
|
97% (341)
|
[STAAB1985]
|
|
28% (105)
|
54% (78)
|
|
95% (150)
|
[SZYMENDERA1 985]
|
|
46% (29)
|
89% (59)
|
|
|
[WANG1985]
|
|
37% (43)
|
68% (44)
|
88% (40)
|
|
[YOSHIKAWA198 5]
|
|
|
|
90% (40)
|
78% (54)
|
[SAFI1987]
|
|
35% (nd)
|
68% (nd)
|
|
|
[GUADAGNI1993 ]
|
|
24% (90)
|
58% (110)
|
|
90% (100)
|
[VONKLEIST1996 ]
|
|
29% (48)
|
52% (71)
|
96% (45)
|
|
Valeurs extrêmes
|
|
11% 940%
|
52% 989%
|
88% 996%
|
70% 997%
|
|
1.6.2. Bilan initial
1.6.2.1. Corrélation avec les stades d'extension
tumorale
Le recherche bibliographique a permis d'identifier huit
études ayant évalués la sensibilité de l'ACE en
fonction des stades d'extension de Dukes. Certaines études
précisent l'amplitude moyenne des concentrations sériques des
marqueurs.
Ces études retrouvent des valeurs extrêmes pour
la sensibilité de 0 et 33% dans les cancers de stade A, de 12 et 49%
dans les cancers de stade B, de 21 et 61% dans les cancers de stade C et de 32
à 86% dans les cancers de stade D.
L'élévation de la concentration sérique de
l'ACE au dessus de la valeur de référence est 3 fois sur quatre
associées à la présence de métastases
viscérales.
L'amplitude moyenne des concentrations observées
augmente avec le stade, elle varie de 2,7 à 4,9g/l dans les stades A, de
4,8à 22,9g/l dans les stades B, de 11.8 à 31,4g/l dans les stades
C et de 39,3 à 475g/l dans les stades D. les écarts types
associés à ces valeurs moyennes sont tels qu'il n'est pas
possible de différencier les stades d'extension sur le base de la
concentration de l'ACE.
La place à accorder au dosage de l'ACE dans le bilan
initial diffère selon les experts et les recommandations
o Pour les experts de la conférence de consensus
organisée en 1998 sous l'égide de l'ANAES, les examens
biologiques, dont le dosage de l'ACE, ne modifient pas l'attitude
thérapeutique. Leurs dosages ne sont pas recommandés
(Anon, 1998).
o Pour les experts de l'ASCO, le dosage de l'ACE est
recommandé s'il peut aider à établir le stade d'extension
ou à prendre la décision opératoire (Anon,
1996).
1.6.2.2. Pronostic
o Données d'ensemble
Les malades ayant une concentration sérique initiale
d'ACE supérieure à la valeur de référence ont un
pronostic plus péjoratif que ceux ayant une concentration normale.
Le pourcentage de récidives est augmenté et/ou
le délai de survenu des ces récidives est plus court.
(Goslin, 1980) ; (Steele, 1982) ; (onetto, 1985) ; (Vang, 1994) ;
(filella, 1994).
L'étude de Landmark, montre que le risque relatif de
décès atteint 5,26 (p0,001) lorsque la concentration
sérique d'ACE dépasse 14g/l pour une valeur de
référence de 2,2 g/l.
o Données de sous groupe
L'extension tumorale pariétale, l'envahissement
ganglionnaire et le nombre de ganglions envahis sont des facteurs pronostiques
standards de survie et/ou de récidive (Adenid, 1998)
o Indépendance par rapport à l'extension
tumorale pariétale
Une étude multi variée montre qu'à tous
les stades de la maladie, chez des patients non métastatiques, la survie
actuarielle diminue si la concentration sérique
préopératoire de l'ACE est supérieure à la valeur
de référence. (Staab, 1981). Mais l'étude
de Carpelin Hallström et celle de Chapman sont en contradiction avec la
première étude et montrent qu'une concentration initiale d'ACE
élevée a une valeur péjorative sur la survie qui est
liée au stade d'extension tumorale. (Carpelan Hallström,
1996A) ; (Chapman, 1998)
Dans l'étude de Behbehani, il n'y a pas de
différence de survie eu niveau de chaque stade en relation avec la
valeur d'ACE sauf dans les stades B1 d'Astler-Coller au moment où la
valeur de l'ACE est supérieure à 20 g/l.
De cela on en conclut que l'indépendance de l'ACE par
rapport à l'extension tumorale pariétale n'est pas
établie.
o Indépendance vis-à-vis
l'envahissement ganglionnaire et du nombre de ganglions
o Patients présentant un envahissement
ganglionnaire
Une étude du gastrointestinal tumor study group
conclut à partir d'une série de 223 patients atteints du cancer
du colon, que des concentrations sériques préopératoires
d'ACE 5 g/l sont associés à un risque significativement plus
élevé de récidives uniquement chez les patients ayant un
à quatre ganglions envahis ( Steele, 1981)
Contrairement, une étude de la Mayo Clinic conclut que
la valeur pronostique de l4ACE n'est vérifiée que si plus de
quatre ganglions sont envahis (Moertel 1986). Cependant un n
après, étudient les mêmes patients en analyse multi
variée, cette équipe retrouve qu'une concentration sérique
préopératoire de l'ACE 10 g/l est un facteur pronostique
indépendant (Scott, 1987)
On ne retrouve aucune corrélation entre la
concentration sérique préopératoire de l'ACE et le nombre
de ganglions envahis dans l'étude publiée par la national
Surgical Adjuvant Breasted Bowel Project (NSABP), ceci est en faveur de
l'indépendance pronostique de l'ACE (Wolmark, 1984).
Lorsque la concentration sérique de
préopératoire de l'ACE est supérieure à la valeur
de référence, le pronostique des patients présentant un
envahissement
ganglionnaire est plus grave. Il y'a une contradiction entre
les résultats prenant en compte le nombre de ganglions envahis.
o Patients sans envahissement ganglionnaire
La recherche bibliographique n'a permis d'identifier que 4
études totalisant 398 patients atteints de cancers de stade B de Dukes
dans les quelles la concentration sérique préopératoire de
l'ACE n'est pas un facteur pronostique de récidive. (Goslin,
1980) ; (Steele, 1982) ; (Moertel, 1986), (Filella, 1994)
Inversement, c'est sur un ensemble de 1259 patients ayant un
cancer colorectal N0, qu'il est démontré que la concentration
sérique préopératoire de l'ACE est un facteur pronostique
indépendant (Staab, 1981) ; (Wolmark, 1984) ; (Wang, 1994) ;
(Landmark, 1995) ; (Harrison, 1997)
o Résultats d'analyses unies
variés
Une étude montre que le pronostic d'un cancer du colon
du stade B de Dukes associé à une valeur
préopératoire de l'ACE élevée est équivalent
à celui d'un cancer de stade C associé à une valeur d'ACE
normale. (Wang, 1994)
o Résultats d'analyses multi
variées
Il a été démontré par deux
études qu'une concentration sériques préopératoire
de l'ACE 5 g/l est un facteur indépendant de pronostic
défavorable sur la survie des patients N0 ayant
bénéficié d'une exérèse complète.
(Staab, 1981) ; (Harrison, 1997)
De ces études on en conclut que l'ACE est un facteur
pronostique indépendant dans les cancers du colon sans envahissement
ganglionnaire, mais reste que son indépendance par rapport à
l'extension tumorale n'est pas prouvée.
1.6.3. Évaluation de l'efficacité du
traitement
Pour pouvoir apprécier sur le plan biologique
l'efficacité d'une thérapeutique, il faut impérativement
mesurer la concentration sérique du marqueur avant traitement.
1.6.3.1. Chirurgie
o Corrélation avec l'efficacité de la
chirurgie
Après exérèse complète de la
tumeur primitive, le normalisation du marqueur est en général
obtenue en 30 jours mais peut prendre jusqu'à 4 mois. (Mach,
1978) ; (Fletcher, 1986)
La constance, six semaines après traitement
chirurgical, de la valeur élevée de l'ACE sérique
détermine la présence de reliquats tumoraux (Anon, 1981)
; (Anon, 1996), ceci s'applique aussi bien à la
résection de cancer primitif qu'à la résection des
métastases. (Mlikacabanne, 1998)
o Valeurs pronostiques sur la survie après
résection des
métastases
Plusieurs études rétrospectives ont
tenté d'individualiser les facteurs pronostiques sur la survie sans
récidive après chirurgie potentiellement curative des
métastases hépatiques (Hohenberger, 1994) ; (Scheele,
1995) ; (Nordlinger, 1996) ; (Wang, 1996) ; (bakalabos, 1999) et
pulmonaires (Baron, 1996) ; (Girard, 1996)
o Métastases hépatiques
Dans trois études, l'analyse unie variée
retient la valeur de l'ACE préopératoire comme facteur de
pronostic sur la survie après chirurgie des métastases, mais en
analyse multi variée, le caractère indépendant n'est pas
démontré. L'étude de Hohenberger montre que le
paramètre le plus significatif c'est l'ACE postopératoire.
(Hohenberger, 1994). La présence de métastases
satellites dans l'étude de Scheele. Dans l'étude d'Adam qui
inclut 64 patients soumis à des métatasectomies hépatiques
itératives, les facteurs pronostiques les plus puissants sont le
caractère curatif de le 2eme hépatectomie et le temps
écoulé entre la 1ere et la seconde métatasectomies.
(Adam, 1997)
Une autre étude mltivariée sur 54 patients,
donne des résultats qui paraissent à première vue
incohérents, l'ACE et le sexe sont les seuls variables ayant une valeur
pronostique indépendante et à une valeur
préopératoire d'ACE 20g/l est associée un taux de survie
meilleur qu'a une valeur 20 g/l. (Wang, 1996).
Ces résultats sont dus à une représentation
importante des tumeurs non sécrétantes.
Une enquête très importante a été
effectuée auprès des membres de l'association française de
chirurgie et elle a concerné 1088 patients opérés, et a
démontré que la survie après résection des
métastases hépatiques est, en analyse mltivariée ,
affectée de façon indépendante par l'âge, la taille
de la plus grande métastase ou par le concentration sérique
préopératoire de l'ACE, le stade de la tumeur primaire, la
durée de l'intervalle libre, le nombre de nodules hépatiques et
les marges de résection. (Nordlinger, 1996)
Une étude rétrospective faite sur 301 patients
établit à 30g/l le seuil de discrimination de l'ACE pour
l'opérabilité des métastases hépatiques et la
survie des patients après chirurgie. (Bakalakos,
1999)
De cela on en conclut que la concentration sérique de
l'ACE mesurée avant hépatectomie a une valeur pronostique sur la
survie des patients opérés, mais dont le caractère
indépendant n'est pas prouvé.
o Métastases pulmonaires
La concentration sérique de l'ACE avant thoracotomie
apparaît dans deux études comme un facteur pronostique sur la
survie des patients chez les quels les métastases pulmonaires ont
été opérées, en analyse uni variées et
(Baron, 1996) et multi variées (Girard, 1996).
1.6.3.2. Réponse à la
chimiothérapie
Si la concentration sérique du marqueur est initialement
élevée, les résultats
des dosages obtenus au cours de la chimiothérapie
constituent un index mesurable de la réponse au traitement et de
l'évolution de la maladie.
o Corrélation avec la réponse au
traitement
Une réponse complète est rarement obtenue avec
la chimiothérapie (Adenis, 1998). Cinq études
ont évalué la valeur prédictive de l'évolution
pré thérapeutique de l'ACE sur une réponse objective
évaluée par les examens cliniques et radiologiques.
(Kouri, 1992) ; (Ward, 1993) ; (Noda, 1996) ; (Hamm, 1998).
o Diminution des concentrations sériques
du
marqueur
La sensibilité de la diminution des concentrations
sériques du marqueur pour une réponse partielle a
été évaluée dans trois études à 84%,
100% et 54%, avec des spécificités respectives de 77%, 65% et 53%
d'où des valeurs prédictives d'une réponse objective de
54%, 67% et 73%. (Kouri, 1992) ; (Ward, 1993) ; (Hamm, 1998).
Dans deux études, la décroissance est significative deux mois
après le début du traitement (Allemmersh, 1987)
; (Noda, 1996), alors que, dans ce même temps,
la réduction du volume tumoral mesurée par scanographie n'est
significative que dans 27% des cas. (Noda, 1996).
o Augmentation des concentrations sériques
du
marqueur
La valeur prédictive de l'élévation de
la concentration sérique de l'ACE sur la progression objective de la
maladie est évaluée à 100% dans l'étude de Ward.
Ces résultats n'ont pas été retrouvés dans la
série de Hamm où la VPP de l'ACE n'est que de 33%. Dans cette
série la variabilité des résultats de dosages est
très importante (26 % d'un jour à l'autre), ce qui peut expliquer
le nombre important de faux positifs observés. (Hamm,
1998).
En début de chimiothérapie, une augmentation
paradoxale et transitoire des concentrations sériques du marquer peut
être observée, elle traduit le relargage du marqueur dans le sang
sous l'effet d'une destruction tumorale importante.
o Discordances
Dans 20% des cas, il existe une discordance entre les
données cliniques, radiologiques et les profils d'évolution du
marqueur. Techniquement, il faut prendre en considération la
différence entre les critères d'évolution biologiques et
les critères d'évolution radio cliniques.
De ce point de vue, il s'agit moins de discordance vraie que
de complémentarité entre plusieurs paramètres de
l'évaluation de l'efficacité du traitement. Ceci démontre
la nécessité de critères précis lorsqu'il s'agit
d'apprécier
une évolution biologique. Les propositions du working
group in tumors markers criteria (Bonfrer, 1990), valables
pour l'interprétation de l'évolution de tous les marqueurs {cf.
SOR biologie marqueurs tumoraux sériques du cancer du sein
(Basuyau, 2000)} sont les suivantes :
o Hors traitement, augmentation régulières sur
trois dosages.
o Sous traitement, progression en cas d'augmentation de plus
de 25%, rémission partielle en cas de diminution de plus de 50%.
Une diminution des valeurs de l'ACE peut être
observée sans réponse tumorale objective. Selon
l'hypothèse physiologique la plus probable, l'action des cytotoxiques
s'exerçant au sein d'une population cellulaire
hétérogène serait plus précoce sur les cellules
sécrétrices que sur les cellules n'exprimant pas le marqueur
(Kouri, 1992) ; (Ward, 1993). Deux études montrent
qu'en cas de non réduction de la taille tumorale, la diminution des
concentrations sériques de l'ACE permet d'identifier un groupe de
patients dont le pronostic est relativement meilleur. (Allenmersh,
1987) ; (Noda, 1996).
Certaines équipes considèrent la
précocité de l'information apportée par le dosage du
marqueur tumoral comme une aide potentielle à la conduite
thérapeutique. Dans une étude pharmacocinétique de
l'administration du 5-FU, il est mis en évidence une relation dose-effet
entre le niveau plasmatique de 5-FU, l'existence d'une « réponse
biologique » et l'intensité de cette réponse (Maillart,
1992). Dans une étude d'escalade de dose d'un inhibiteur métallo
protéinases, il est mis en évidence une relation entre la dose
d'inhibiteurs administrés et l'intensité de la réponse
biologique. (Primrose, 1999)
Une augmentation progressive des concentrations
sériques du marqueur est le signe de résistance au traitement.
Malgré la rareté des publications sur ce sujet, il existe un
consensus entre les membres de l'ASCO pour affirmer que la valeur
prédictive d'une augmentation de l'ACE est suffisamment
élevée pour écarter la nécessité d'une
confirmation radiologique. (Anon, 1996)
Toujours selon l'ASCO, l'augmentation de la concentration
sérique du marqueur observée sur deux prélèvements
est une indication suffisante de progression de la maladie et incite à
interrompre le traitement même en l'absence de confirmation clinique ou
radiologique. (Anon, 1996)
o Valeur pronostique
o Valeur pronostique de décroissance
Plusieurs études effectuées chez des patients
traités par chimiothérapie intra hépatique
démontrent que la survie est corrélée à
l'intensité de la décroissance de l'ACE pendant le traitement
et/ou la durée pendant la quelle l'ACE se maintient en dessous des
valeurs de références (Allenmersh, 11987) ; (Quentmeier,
1989) ; (Node, 1996) ; (Hamazaki, 1998). Cependant, aucune
étude prospective intégrant la décroissance de l'ACE comme
paramètre du suivi thérapeutique n'a été
relevée dans la littérature.
o Valeur pronostique de l'ACE avant
chimiothérapie
Une étude rétrospective compilant les
résultats obtenus par le Hellenic Cooperative Oncology Group lors
d'essais comparatifs de protocoles de chimiothérapies à base de
5-FU-acide folinique montre que parmi les paramètres biologiques, les
valeurs pré thérapeutiques de la ãGT, de l'albumine et de
l'ACE sont, en analyses multi variées, des facteurs indépendants
sur la survie (Fountzilas, 1996)
Deux études rapportant les travaux du Royal Marsden
Hôpital ont mis en évidence la valeur pronostique de la
concentration sérique pré chimiothérapique de l'ACE.
(Webb, 1995) ; (Asserdon, 1999).
Une analyse multi variée effectuée en 1995 pour
évaluer la valeur pronostique de plusieurs marqueurs tumoraux, montre
que l'ACE est un facteur pronostique sur la survie qui est indépendant
de l'indice de performance et de la différenciation cellulaire
(Webb, 1995). Une étude multiparamétrique parue
en 1999, incluant 497 patients dans un essai comparatif de voies
d'administration du 5-FU, montre qu'a une concentration sérique initiale
d'ACE égale ou supérieure à 5tg/l est associée une
probabilité réduite de répondre au traitement, notamment
en ce qui concerne les cibles locales ou ganglionnaire où la
possibilité de réponse est divisée par 4 en cas d'ACE
élevée. Dans cette étude, la concentration sérique
de l'ACE mesurée avant chimiothérapie est un facteur pronostique
majeur sur la survie globale, comparable à l'indice de performance et
à l'administration du 5-FU en boulus. (Asserohn,
1999)
La concentration sérique initiale de l'ACE
mesurée avant chimiothérapie est un facteur pronostique
indépendant sur la survie des patients, mais si la
concentration d l'ACE augmente en début de la
chimiothérapie cela traduit une progression de la maladie.
Enfin le dosage de l'ACE peut être
complémentaire de l'imagerie et de l'examen clinique pour
apprécier la réponse à la chimiothérapie. Il est
particulièrement indiqué qu'en ca de maladie non mesurable.
1.6.4. Surveillance des patients traités par
chirurgie
curative
Vingt à trente pour cent des malades ayant
bénéficié d'une chirurgie curative récidivent
après deux ou trois ans. Le premier site de dissémination par
voie hématogène est le foie, dans 40% des cas c'est le seul site
atteint, en deuxième position vient le poumon. Les autres organes sont
rarement atteints en l'absence d'atteinte hépatique ou pulmonaire. Les
récidives ganglionnaires rétro péritonéales sont
observées dans 10% des cas. Les récidives anastomotiques sont
très rares 6à8% et très souvent associés à
une dissémination péritonéale et/ou métastatique.
(Cohen, 1993) ; (Niederhunber, 1993) ; (Obrand, 1997)
Moins de 5% des malades sont survivants à 5 ans, en
absence d'un traitement spécifique.
Le seul traitement potentiellement curatif est chirurgical :
o Rapporté à l'ensemble des patients atteints
de cancer coloréctal, le bénèfice de la résection
des métastases est très faible, 1 à 5% d'augmentation de
la survie à 5 ans. (Northover, 1985) ; (Cohen, 1993) ; (Wolf,
1997)
o Le bénéfice est significatif eu niveau
individuel, 20% des patients atteints de récidive hépatique ou
pulmonaire peuvent bénéficier de résections curatives avec
une survie à 5 ans estimée entre 23% et 40% (Niederhuber,
1993)
Le recours à la chimiothérapie reste possible,
en cas d'impossibilité de résection. Dans l'essai clinique du
Nordic Gastrointestinal Tumor Adjuvent Therapy Group, la survie étant
d'autant meilleurs que le traitement est précoce, délivré
alors que les patients sont encore asymptomatique. (Anon,
1992).
1.6.4.1. ACE indicateur de récidive
Dans plus de 65% des cas, l'augmentation des concentrations
sériques de l'ACE est le premier indicateur de récidive ; elle
précède l'apparition de signes cliniques ou radiologiques de
reprise évolutive avec une avance au diagnostic estimé entre 4 et
12 mois. (Mach, 1978) ; (Tate, 1982) ; (Sugarbaker, 1987) ; (Wanebo,
1989) ; (Mc call, 1994) ; (Wolf, 1997).
1.6.4.2. Sensibilité et spécificité
de l'ACE pour la détection des récidives
Une concentration sérique de l'ACE supérieure
à la valeur de référence a une valeur prédictive
pour la détection des récidives d'environ 80%. Dans le but de
réduire le cout de la surveillance, une étude suédoise
(Graffner, 1985), a comparé l'efficacité de
plusieurs outils diagnostiques pour la détection des récidives
des cancers colorectaux. Un suivi clinique traditionnel a été
comparé à un suivi clinique traditionnel comportant en plus une
surveillance intensive, comprenant les dosages de l'ACE, de la PA, de la GT, de
l'hémoglobine, la mesure de la vitesse de sédimentation des
érythrocytes et l'électrophorèse des protides.
La valeur prédictive positive de l'ACE est très
supérieure à celle des autres tests biologiques (VPP de l'ACE
79,4%, VPP des autres tests biologiques est de 5 ,5% à 13,8%) avec une
sensibilité de 89% pour la détection des récidives
hépatiques. Par contre, en cas de résultat normal, tous les tests
biologiques ont une forte valeur prédictive pour éliminer une
atteinte secondaire (les VPN de l'ACE de la PA, de la GT sont respectivement
98,7%, 96%, 87,6%). Les auteurs ont conclus que l'ACE devrait être le
seul parametre biologique dans de le suivi des cancers colorectaux.
(Graffner, 1985).
La valeur de l'ACE pour la détection des récidives
d'un cancer colorectal a été évaluée en fonction
:
· Du caractère sécrétant de la tumeur
primitive
· Du site de la récidive
· De la méthode d'appréciation des
augmentations des concentrations sériques de l'ACE.
o Valeur pronostique en fonction du caractère
sécrétant
de la tumeur primitive
Il est très rare que des cancers à concentration
initiale d'ACE élevée rechutent sans augmentation du marqueur. La
sensibilité du test est supérieure si la tumeur primitive
sécrète l'ACE, mais il existe des tumeurs non
sécrétantes initialement dont la rechute sera marquée par
des valeurs sériques d'ACE supérieurs à la valeur de
référence.
Dans une étude, au seuil de 5tg/l, la
sensibilité de l'ACE pour le détection des rechutes qui est de
97% ( spécificité 88%) en cas d'ACE préopératoire
supérieur à la valeur de référence, n'est plus que
de 66% ( spécificité 94%) en cas d'ACE
préopératoire inferieur à la valeur de
référence. (Wang, 1994).
Dans deux études menées chez des patients ayant
une tumeur de stade C de Dukes initialement non sécrétant, la
sensibilité d'une valeur sérique de l'ACE supérieure
à la valeur de référence pour la détection des
récidives est estimée à 36% (spécificité
93%) et 44%. Chez ces patients, l'augmentation de la concentration
sérique du marqueur est pratiquement toujours associée à
l'apparition de métastases viscérales. (Zeng, 1993) ;
(Tobaruela, 1997).
Pour certains auteurs, les tumeurs peu
différenciées peuvent être suivies par le dosage de l'ACE
si le marqueur est détecté par immunohistochimie dans la tumeur
primitive. (Goslin, 1981) ; (Zeng, 1993).
La valeur diagnostique de l'Ace varie en fonction du
caractère sécrétant ou non de la tumeur mais un taux
initial normal ne doit jamais exclure les marqueurs des paramètres de
surveillance.
o Valeur pronostique en fonction du site de
récidive
La sensibilité de l'ACE est d'environ 78% à 97%
pour la détection des récidives intra hépatiques , de 75%
pour les récidives rétro péritonéales, de 45% pour
celles qui sont locorégionales et de 42 à 50% pour les
métastases pulmonaires (Moertel, 1993) ; (Bergamashi,
1996).La sensibilité de l'ACE diminue en cas de
métastases solitaires, dans l'étude de Moertel, elle est de 76%
pour les métastases hépatiques solitaires alors que pour les
métastases pulmonaires solitaires elle n'est que de 15%.
La valeur diagnostic de l'ACE varie en fonction du site de la
récidive. La sensibilité de l'ACE pour la détection des
récidives est meilleure lorsque les récidives sont
hépatiques que lorsqu'elles atteignent d'autres sites.
o Valeur pronostique en fonction de la
méthode
d'appréciation des augmentations de
l'ACE
Plusieurs méthodes d'appréciation des
augmentations des concentrations sériques de l'ACE ont été
décrites. Elles peuvent être réparties en deux groupes,
groupe des méthodes comparant le résultat à une valeur
seuil et groupe utilisant la cinétique d'évolution des
concentrations sériques de l'ACE.
o Méthodes comparant le résultat à
une valeur seuil
Selon les pratiques, le seuil retenu pour la surveillance est
un seuil individuel ou un seuil fixé pour l'ensemble des patients
à partir de la valeur de référence (Tableau IV),
(Lucha, 1997).
Tableau IV : études ayant
évalué la valeur diagnostique de l'ACE en fonction d'une
méthode basée sur l'adoption d'une valeur seuil. (Lucha,
1997)
|
Références
|
Effectif
suivi
|
Nombre
de
récidives
|
Valeur de
référence(ug.L-
1)
|
Valeur
Seuil(ug.L-
1)
|
Sensibilité
%
|
Spécificité
%
|
VPP
%
|
VPN
%
|
|
KOCH1982
|
181
|
35
|
5
|
10
|
31
|
96
|
65
|
85
|
|
LUNDE1982
|
179
|
44
|
3,5
|
3,5
|
75
|
78
|
46
|
92
|
|
STEELE1982
|
405
|
134
|
nd
|
2,5
|
90
|
31
|
33
|
96
|
|
242
|
102
|
5
|
68
|
64
|
42
|
84
|
|
128
|
80
|
10
|
54
|
88
|
62
|
83
|
|
83
|
56
|
20
|
38
|
93
|
67
|
80
|
|
SZYMENDERA1982
|
143
|
13
|
3,2
|
3,2
|
|
|
|
91
|
|
169
|
15
|
|
7,5
|
91
|
|
TATE1982
|
468
|
83
|
20
|
40
|
65
|
91
|
69
|
90
|
|
CARLSSON1983
|
139
|
50
|
3
|
3
|
90
|
66
|
60
|
92
|
|
|
|
7,5
|
78
|
91
|
83
|
69
|
|
GRAFFNER1985
|
190
|
47
|
nd
|
Valeur de
référence
|
66
|
99
|
79
|
99
|
|
DENSTMAN1986
|
176
|
55
|
3
|
4
|
47
|
81
|
53
|
77
|
|
nd
|
nd
|
(non fumeurs)
|
5
|
60
|
73
|
50
|
80
|
|
214
|
76
|
5
|
6
|
62
|
83
|
67
|
80
|
|
176
|
55
|
(fumeurs)
|
7,5
|
55
|
88
|
68
|
81
|
|
176
|
55
|
|
10
|
38
|
93
|
70
|
77
|
|
176
|
55
|
|
20
|
31
|
98
|
85
|
76
|
|
MOERTEL1993
|
1017
|
417
|
5
|
3
|
73
|
62
|
57
|
77
|
|
417
|
5
|
59
|
84
|
72
|
75
|
|
417
|
10
|
45
|
96
|
89
|
72
|
|
417
|
15
|
36
|
99
|
96
|
69
|
|
MCCALL1994
|
311
|
98
|
5
|
5
|
58
|
93
|
79
|
83
|
|
WANG1994
|
272
|
27
|
5
|
5
|
83
|
93
|
66
|
94
|
|
LUCHA1997
|
280
|
44
|
5
|
5
|
67
|
92
|
71
|
90
|
|
Valeurs extrêmes
|
|
|
|
|
31-90
|
31-99
|
33-96
|
69-99
|
NB : nd= non défini
L'interprétation des résultats de dosage de
l'ACE en fonction d'un seuil individuel est le fait d'une équipe. Le
seuil individuel est déterminé à partir de la ligne de
base des concentrations de l'ACE post opératoire. Les augmentations de
l'ACE sont interprétées avec un intervalle de confiance
analytique de 95%. La valeur prédictive de cette méthode sur la
découverte chirurgicale d'une récidive est de 88% dans la
population initiale (Martin, 1977), mais le nombre important
de faux positifs
dus aux fluctuations individuelles en a limité la
diffusion (Rittgers, 1978). La majorité des auteurs
utilisent un seuil qui est égal à la valeur de
référence ou à un multiple de cette valeur (Koch,
1982) ; (Lunde, 1982) ; (Szymendera, 1982) ; (Tate, 1982) ; (Carlson, 1983) ;
(Graffner ; 1985) ; (Moertel, 1993) ; (Mc call, 1994) ; (Wang, 1994) ; (Lucha,
1997).
o Lorsque le seuil est égal à la valeur de
référence, la sensibilité pour la détection de
récidive varie de 58% pour une spécificité de 93% à
90% pour une spécificité de 66%, la VPP varie de 41% à 79%
et la VPN de 75% à 99%.
o Lorsque le seuil est égal au double de la valeur de
référence, la sensibilité pour la détection de
récidive varie de 31% pour une spécificité de 96% à
78% pour une spécificité de 91%, la VPP varie de 65% à 89%
et la VPN de 69% à 90%.
L'analyse des études évaluant la
fiabilité du test à plusieurs seuils d'interprétations
montrent que l'adoption d'un seuil élevé permet
d'améliorer la valeur prédictive du test, mais au prix d'une
diminution importante de sa sensibilité (Steele, 1982) ;
(Carlsson, 1983) ; (Denstman, 1986) ; (Moertel, 1993).
o Méthodes utilisant une appréciation
dynamique des
concentrations sériques de l'ACE
Plusieurs auteurs ont plaidé très tôt pour
une appréciation dynamique des variations des concentrations
sériques d'ACE, plus sensible que le seuil puisqu'elle permet de
constater l'élévation du marqueur même dans la zone des
valeurs normales, et plus spécifique puisqu'elle permet
d'éliminer les variations transitoires des concentrations
sériques du marqueur (Rittgers, 1978) ; (Staab, 1978). (Tableau
V), (Mora, 1997).
Tableau V : études ayant évalué la
valeur diagnostique de l'ACE en fonction
d'une méthode basée
sur l'appréciation dynamique des variations des
concentrations
sériques. (Mora, 1997)
|
Références
|
Effectif
|
Nombre
de
récidives
|
Type de cinétique
|
Sensibilité
%
|
Spécificité
%
|
VPP
%
|
VPN
%
|
|
STEELE1982
|
456
|
112
|
augm de 3% par mois
de log (1+ACE)
|
63
|
90
|
66
|
88
|
|
SZYMENDERA1982
|
231
|
75
|
augm par rapport à
ligne de
base
postopératoire (sans
autre précision)
|
83
|
99
|
97
|
86
|
|
BOEY1984
|
146
|
51
|
augm de 5% par mois
de log (1+ ACE)
|
86
|
76
|
66
|
71
|
|
DENSTMAN1986
|
150
|
37
|
augm de 12,6%par
mois de ACE
|
62
|
95
|
79
|
88
|
|
SUGARBAKER1987
|
66
|
33
|
3 valeurs>ligne de
base
postopératoire,en
augmentation,au moins
une
valeur>1ug.L-1
|
81
|
94
|
93
|
85
|
|
MORA1997
|
75
|
18
|
augm par rapport à
ligne de
base
postopératoire (analyse
de variance)
|
88
|
96
|
94
|
96
|
|
NAKAYAMA1997
|
264
|
39
|
pente d'augmentation>0
|
79
|
88
|
54
|
96
|
Pour certains auteurs, l'analyse cinétique se
résume à la mise en évidence sur plusieurs
prélèvements successifs d'une augmentation de la concentration de
l'ACE par rapport à la ligne de base postopératoire
(Szymendera, 1982) ; (Sugarbaker, 1987) ; (Mora, 1997) ; (Nakayama,
1997).
Pour d'autres, l'analyse se pente intervient après
transformation logarithmique log (1+ACE), ce qui met en évidence le
caractère exceptionnel de l'augmentation du marqueur (Steele,
1982) ; (Boey, 1984) ; (Denstman, 1986), tab. Les malades qui
demeurent en rémission clinique ont une pente d'évolution du
marqueur sensiblement nulle.
En cas de récidive, la valeur médiane de
l'accroissement mensuel est de 5,8% dans l'étude de Steele et de 20%
dans l'étude prospective de Boey. ( 20% de récidive en cas de
récidive versus 0,3% en l'absence de récidive, p0,001).
Une étude utilise un modèle mathématique
tenant en compte de la croissance exponentielle de la tumeur, de la dilution du
marqueur dans le compartiment vasculaire et de la contribution à la
production du marqueur par le tissu normal. La sensibilité et la
spécificité ne sont pas plus élevées que dans une
approche cinétique plus simple (Carl, 1993). L'analyse
de pente permet le calcul du temps de doublement du marqueur.
Beaucoup d'autres s'accordent sur le fait que la perte
d'accroissement est plus rapide en cas de métastase hépatique
qu'en cas de récidive locale et que le temps de doublement pourrait
constituer une aide pour la localisation des récidives (
Steele,1982) ; (Boey, 1984) ; (Carl,1993) ; (Umehara, 1993).
Le temps de doublement du marqueur précédent le
diagnostic de la rechute est un facteur pronostique sur la survie, une
augmentation rapide des concentrations sérique du marqueur ayant une
signification péjorative. (Staab, 1985) ; (Koreyema,
1997).
Deux études comparant chez les mêmes patients
l'efficacité de la comparaison, des résultats à
différents seuils s et de l'analyse dynamique montrent que le meilleur
accord entre la précision biologique de la récidive et la
réalité est obtenue avec un seuil un peu supérieur
à la valeur de référence mais que cet accord est inferieur
à celui obtenu avec une analyse dynamique. (Steele, 1982)
;(Denstman, 1986).
En conclusion :
o L'ACE est le premier indicateur de récidive dans 65%
des cas.
o L'ACE est le marqueur de choix pour surveiller les patients
atteints de cancer colorectal.
o Il est très rare que des cancers à concentration
sérique initiale
d'ACE élevée rechutent sans augmentation du
marqueur.
o Il n'y a pas de méthodes standard pour apprécier
l'augmentation
des concentrations sériques d'ACE dans le cadre de la
surveillance des patents après chirurgie curative.
1.6.4.3. Intérêt de la
surveillance
Une surveillance biologique soutenue incluant le dosage de l'ACE
permet de prédire la survenue de récidive avec une avance de
quelques semaines à quelques
mois sur le diagnostique clinique ou radiologique, cette
surveillance permet aussi le diagnostic de récidives à un stade
où l'opérabilité est meilleure. Mais il n'existe pas
à l'heure actuelle, à l'échelle d'une population de
malades des suivis, de bénéfices démontrés, en
temps de survie, résultant de cette avance au diagnostic et cette
meilleure opérabilité. L'inclusion du dosage de l'ACE dans la
surveillance des cancers du colon varie selon les comités d'experts :
o Pas de surveillance se l'ACE en dehors d'essais
thérapeutiques
o Surveillance régulière de l'ACE chez les patients
en état de supporter une thérapeutique autre que
symptomatique.
o En l'absence de consensus, la surveillance
régulière des concentrations de l'ACE après chirurgie
curative n'est pas recommandable systématiquement.
La place du dosage de l'ACE dans le suivi des patients devra
être évaluée à la publication études et au
moment où des nouvelles techniques d'imagerie tel que le scanner
hélicoïdal et de chirurgie tel le traitement percutané des
métastases seront plus largement utilisées et que le traitement
systématique du cancer aura gagné en efficacité.
2. Implication de la protéine â
caténine-APC dans le développement du cancer du colon
2.1. Gène APC
Localisé sur le bras long du chromosome 5,
possédant 15 exons et codant pour une protéine de 311kda,
constituée de 2844 acides aminés formant des homodiméres
par sa partie aminoterminale. Elle est localisée au pole
basolatéral de la cellule épithéliale colique. Son
expression est d'autant plus importante que la cellule migre du fond de la
crypte vers le sommet de la villosité. Outre la béta
caténine, quatre partenaires de la protéine APC sont maintenant
connus et forment avec elle des complexes protéiques. Il s'agit de la
protéine EB1 de 30kda, dont la fonction est inconnue (Su et
a!, 1995), la protéine DLG codée par un
gène humain homologue du gène suppresseur de tumeur chez la
drosophile « Disk large » (Matsumine et a!,
1996), de la glycogen synthase kinase 3béta
(Rubinfield et al, 1996) et des microtubules composants du
cytosquelette (Smithe et a!, 1994). Bien que
l'interaction de la protéine APC avec EB1 et DLG soit probablement
importante dans le processus de transformation maligne (puisque la plupart des
altérations du gène APC dans les cellules tumorales coliques
conduisent à la synthèse d'une protéine tronquée ne
possédant pas ses domaines de liaisons), les avancées
récentes sur la compréhension du rôle de la protéine
APC sont venues de l'étude de la béta caténine. En effet,
son interaction avec cette protéine semble être
l'élément qui fait le lien entre deux processus apparemment
différents ; l'adhésion et la prolifération cellulaire.
(fig 12) (Rubinfield et a!, 1996).

Figure 12: structure du gène APC (Rubinfield et
a!, 1996)
2.1.1. Mutations du gène APC et instabilité
chromosomique
L'analyse génétique des cancers colorectaux a
permis de révéler l'existence de deux formes distinctes. La
première retrouvée dans 15% des cancers, est associée
à un défaut du système de réparation des
mésappariements de l'ADN qui se traduit par une instabilité des
microsatellites (Phénotypes MSI+ ou RER+) (Puig et al, 1999).
La seconde, de loin la plus fréquente, puisque retrouvée
dans 85% des cancers colorectaux, se caractérise par une
instabilité chromosomique (Phénotype LOH+). L'origine de cette
instabilité chromosomique n'était jusqu'à présent
pas clairement identifié, mais deux articles récents montrent que
les mutations de la protéine APC pourraient bien en être
responsables.
Deux équipes décrivent en effet dans Nature
Cellul Biology que des cellules embryonnaires de souris homozygotes pour la
mutation Min du gène APC acquièrent, au cours des passages en
culture, des anomalies du nombre de chromosomes qui sont liées à
un défaut de ségrégation des chromosomes. (Fodde
et a!, 2001) ; (Kaplan et a!, 2001).
L'allèle Apc min code pour une protéine
tronquée de sa partie carboxyB terminale, qui ne peut alors ni se lier
aux microtubules, ni participer à la dégradation de la
béta caténine. D'autres mutations, comme la mutation Apc1638T,
située dans une région plus distale, n'altèrent pas la
fonction de dégradation de la béta caténine, mais
provoquent aussi une instabilité chromosomique. Ceci suggère que
cette instabilité n'est pas liée à un effet
tanscriptionnel induit par le complexe béta caténine/TCF4.
(Kaplan et a!, 2001). Les deux groupes observent que
la protéine APC native est localisée, pendant la mitose, à
l'extrémité positive des microtubules, au niveau des
kinétochores. Il s'agit probablement d'un attachement physique de la
protéine APC à l'extrémité des microtubules, car
leur dépolarisation empêche cette localisation. La protéine
mutée Apc min est incapable de se lier correctement aux
kinétochores. Il existe alors une désorganisation du fuseau
mitotique, de nombreux microtubules ne s'attachent plus aux
kinétochores, et on observe parfois des
centrosomes surnuméraires, ces anomalies étant
très certainement à l'origine de l'instabilité
chromosomique.
L'interaction entre les microtubules et la protéine Apc
se ferait par l'intermédiaire de la protéine EB1, dont la
localisation normale au niveau des kinétochores disparaît dans les
cellules exprimant la protéine Apc mutée. (Fodde
et al. 2001). Les deux équipes
émettent l'hypothèse d'un rôle de la protéine Apc
dans la stabilisation des microtubules et leur attachement aux chromosomes.
En outre, Apc interagit avec d'autres protéines du
kinétochore, dont la protéine kinase BUB1qui intervient dans le
point de contrôle mitotique. (Kaplan et al,
2001), cependant la signalisation de cette interaction n'est pas
connue.
La fonction de la protéine Apc n'est donc pas
restreinte à celle de gène suppresseur de tumeur, mais pourrait
aussi être essentielle à une ségrégation correcte
des chromosomes lors de la mitose. Son altération pourrait aussi rendre
compte de l'aneuploïdie observée lors de la transformation maligne
des cellules.
2.1.2. Le gène APC, un partenaire essentiel de la
voie Wnt
La protéine Apc joue un rôle majeur dans le
contrôle de l'activité de la voie de signalisation Wnt. Cette voie
aboutit à la formation du complexe de transactivation béta
caténine /TCF4, qui active la transcription de nombreux gènes
cibles, en particulier l'oncogène C-Myc ou la cycline D1. La
protéine Apc favorise la dégradation de la béta
caténine au sein d'un complexe contenant aussi l'axine et la
glycogène synthase kinase 3béta. La principale conséquence
des mutations inactivatrices d'Apc est l'accumulation de la béta
caténine dans le cytosol et le noyau et donc l'activation de la voie
Wnt. S'il est bien établi qu'Apc a un rôle de gène
suppresseur de tumeur, il semble cependant que ce ne soit pas sa seule
fonction. En effet la protéine Apc peut se lier par sa partie
carboxy-terminale, aux microtubules soit par l'intermédiaire de EB1, une
protéine qui est elle-même associée à
l'extrémité positive des microtubules, ceci suggérait que
Apc puisse jouer un rôle dans l'organisation du cytosquelette ou dans
celle du fuseau mitotique. (He et al. 1998).
2.2. Caractérisation d'une voie importante dans la
signalisation de la carcinogénèse colorectale : voie
Wnt
2.2.1. Signalisation Wnt/
â-caténine
La voie de signalisation Wnt / wingless joue un rôle
important au cours du développement embryonnaire en modulant
l'expression des signaux intercellulaires qui contrôlent la croissance,
la migration, le déterminisme et la polarisation cellulaire.
(Bellaiche et perrimon, 1997). Elle a été
particulièrement étudiée au cours du développement
chez le nématode, la drosophile et les amphibiens, mais ses constituants
sont très conservés chez les vertébrés. Dans les
tissus adultes, elle participe au contrôle de la prolifération
cellulaire. Une activation anormale de cette voie par mutation de l'un ou
l'autre de ses composants joue également un rôle important dans un
grand nombre de cancers humains. (Romagnolo, 1997). Cette voie
est alors appelée souvent voie APC/béta caténine/TCF.
Que ce soit au cours du développement ou de la
carcinogenèse, la clé de voûte de cette voie est la
béta caténine ; lorsque la voie est inactive, celle-ci est
dégradée, lorsque la voie est active, elle est
libérée dans le cytoplasme où elle exerce ses fonctions.
(Romagnolo, 1997). Dans la position « off », la
béta caténine est partie intégrante d'un complexe
multiprotéique constitué de l'axine (ou son homologue le
conductine), de la protéine phosphatase PP2A, de la glycogen synthase
kinase 3béta et d'Apc. Au sein de ce complexe l'interaction entre
l'axine et GSK3béta semble favoriser la phosphorylation de la
béta caténine et sa dégradation ultérieure par le
protéasome. La position « on » est obtenue de manière
différente selon qu'il s'agit d'un processus normal du
développement ou d'un processus de carcinogenèse. Dans le premier
cas, l'activation des récepteurs Frizzled par la fixation de Wnt conduit
à la phosphorylation de la protéine Dishevelled qui, par son
association à l'axine, empêche la GSK3béta de phosphoryler
ses substrats parmi lesquels se trouve le béta caténine, rendant
ainsi la liberté à cette dernière. Dans les cancers,
indépendamment de toute activation du récepteur Frizzled, c'est
l'inactivation ou l'activation de gènes codants pour des
protéines impliquées dans le complexe phosphorylant la
béta caténine qui bloque la dégradation de béta
caténine. Le plus souvent, il s'agit de mutation d'Apc mais il peut
aussi s'agir de mutation au niveau de séquences
répétées codantes du gène codant pour
l'axine, ou celles activatrices, dans l'exons 3 du gène codant pour la
béta caténine au niveau des sites de phosphorylation.
Dans tous les cas, lorsqu'elle n'est pas
dégradée, la béta caténine s'accumule dans le
cytoplasme, une partie a un rôle dans l'adhérence cellulaire en
s'associant avec les E-cadhérines et l'actine du cytosquelette pour
former les fonctions gap. Une autre partie de la béta caténine
libre est véhiculée dans le noyau où elle s'associe aux
facteurs de transcription TCF/LEF, modulant ainsi leur activité et
activant l'expression d'un grand nombre de gènes cibles (Fig13)
, (Behrens et al, 1996) ; (Korinek et al, 1997).

Figure 13: représentation schématique des
différents partenaires
de la béta caténine (Korinek et al,
1997).
2.2.2. La â caténine l'élément
clé dans les mécanismes de régulation de la voie
Wnt
Il y'a peu de temps encore, on limitait le rôle de la
béta caténine et son homologue Armadillo, chez la drosophile, aux
processus d'embryogenèse et aux mécanismes cellulaires permettant
l'adhésion des cellules entre elles. (Peifer et al,
1997) ; (Resnik et al, 1997). En effet la béta
caténine est d'une part, l'un des éléments du signal de
transduction qui participe au développement axiale de l'embryon, et
d'autre part, l'un des composants des jonctions adherens. (Heasman et
al, 1994) ; (Kemler et al, 1993).
Plus récemment l'étude de la cancérogenèse colique.
En particulier, l'intérêt qui lui a été porté
dans la carcinogenèse colique a permis de montrer que la béta
caténine semble être un acteur majeur de la transformation maligne
d'une cellule épithéliale colique.
En particulier, l'intérêt qui lui a
été porté dans la carcinogenèse colorectale a
commencé à prendre de l'ampleur lorsqu'elle a été
reconnue comme l'un des partenaires cellulaires de la protéine Apc.
(Su et al, 1993) ; (Rubinfield et al,
1995).
Les mutations du gène de la béta caténine
affectent la région aminoterminale de la protéine la rendant
réfractrice à sa régulation par APC.
La béta caténine n'est pas essentiellement dans le
colorectal mais aussi dans de nombreux autres cancers. (Piard et
al, 2002).
2.2.2.1. Structure de la â
caténine
Trois domaines importants ont été identifiés
sur la béta caténine
o Le domaine N-terminale, composé de 149 acides
aminés, possédant de nombreux sites de phosphorylation par
GSK-3b, par la caséine Kinase (CKIa) et un site de fixation
(sérine 33/37) à la protéine b-TrCP initiatrice du
processus de dégradation par le protéasome.
o Domaine central comprenant 12 ARD (Armadillo Repeat Domain)
de 42 acides aminés chacun. Reconnaissant notamment la protéine
Apc, l'axine/conductine ou encore les facteurs de transcription de la famille
TCF.
o Un domaine C-terminal de 108 acides aminés ayant une
fonction, lui permettant de jouer au niveau nucléaire son rôle
d'activateur de la transcription (Fig 14.) (Peifer, 1997).

Figure14: structure de la â caténine
(Peifer, 1997). 2.2.2.2. Rôle de la â caténine
Dans une cellule normale et en l'absence des facteurs de la
famille wnt la concentration de béta caténine est maintenue
à un niveau très faible par une dégradation constitutive
de la protéine par le protéasome. (Aberle et a!,
1997). Dans les processus de développement, les facteurs
de la famille wnt induisent, par l'intermédiaire de leurs
récepteurs membranaires Frizzled et l'activation de la protéine
cytoplasmique intermédiaire Dishevelled, une déstabilisation de
ce complexe en inhibant l'activité de la GSK3béta.
L'élévation du niveau de béta
caténine libre cytoplasmique dans les cellules tumorales coliques est
suivie de la translocation nucléaire de la protéine. La
béta caténine ne possédant pas de séquence de
nucléarisation, pourrait être transloquée vers le noyau en
s'associant aux facteurs Tcf/Lef via son domaine ARD. (Behrens et al,
1996) ; (huber et a!, 1996). Cette translocation peut
cependant être indépendante des facteurs Tcf/Lef. (Fogotto
et a!, 1998). Dans le noyau, le complexe béta
caténine/Tcf-Lef se lie à des gènes cibles par
l'intermédiaire du domaine de liaison à l'ADN des facteurs
Tcf/Lef, alors que les domaines d'activations transcriptionnelle sont
apportés par la béta caténine. Dans les cellules coliques
présentant des mutations d'APC ou de béta caténine, ce
complexe active de façon constitutive la
production de gènes dont les produits contribuent à
la progression tumorale. (TableauVI)
C-Myc est un facteur de transcription oncogénique dont
les gènes cibles produisent des protéines impliquée,
notamment dans le régulation du cycle cellulaire et de l'apoptose. Le
gène C-Myc a récemment été identifié comme
une cible directe du complexe de la béta caténine/Tcf-Lef.
(He et a!, 1998). Son promoteur contient des
séquences consensus de liaison à l'un des facteurs de la famille
Tcf, « cf-4 binding élément ». Il est surexprimé
dans les cellules tumorales coliques présentant une mutation d'APC ou de
béta caténine. L'activation incontrôlée du complexe
transcriptionel béta caténine/Tcf-Lef expliquerait donc la
surexpression de C-Myc très fréquemment dans les formes
héréditaires ou sporadiques des cancers colorectaux.
La cycline D1, un autre régulateur important du cycle
cellulaire, a été identifié comme une cible directe du
complexe béta caténine_Tcf. (Shtutman et a!,
1999). Cela expliquerait sa surexpression dans 30% des
adénocarcinomes et polypes adénomateux du colon.
La matrilysine est une protéine de la famille des
métalloprotéinases. Elle est impliquée dans les processus
de dégradation de la matrice extracellulaire nécessaire à
l'invasion tumorale et la formation e métastase. Deux sites de liaison
à Tcf-4 ont été identifiés sur son promoteur et sa
transcription est activée par le complexe béta
caténine-Tcf-4 (Crawford et a!, 1999). La
matrilysine est surexprimée dans 80% des adénomes et des
carcinomes coliques humains résultant de la mutation
générale ou somatique d'APC. Son expression est totalement
corrélée à celle de la béta caténine dans
les tumeurs. Elle est également retrouvée dans les polypes
précancéreux, contrairement aux autres
métalloprotéinases qui sont exprimées tardivement au cours
de la carcinogenèse.
La gastrine, puisant stimulant de la sécrétion
acide gastrique, est également un facteur de croissance pour les
cellules tumorales coliques. Les produits issus s'une mutation
post-traductionnelle incomplète de l'hormone surexprimés dans les
cancers du colon, jouent un rôle important dans la prolifération
des cellules normales ou tumorales de la muqueuse colique. (Van Solinge
et a!, 1993) : (Wang et a!, 1996) ; (Koh et a!,
1999). L'analyse du gène de la gastrine a
révélée l'existence de plusieurs
sites de liaisons potentiels du facteur Tcf. (Koh et
a!, 2000). Sur des cellules en cultures, l'expression d'une
forme constitutivement active de béta caténine augmente
l'expression du gène de la gastrine. Inversement, la transfection d'un
mutant dominant négatif TCF4 inverse cet effet. L'activation du
gène de la gastrine par le complexe béta caténine-tcf4
pourrait donc contribuer à la progression tumorale des cellules
coliques.
Il semble claire que la béta caténine puisse
être définie comme un oncogène jouant un rôle
important dans la progression tumorale colique. Elle est constitutivement
activée très précocement au cours de le
carcinogenèse par les altérations géniques qui peuvent
soit l'affecter directement, soit touché des protéines
impliquées dans la régulation de sa dégradation tel que
APC, GSK3béta. Son activation constitutive conduit à l'expression
dérégulée des gènes cibles impliqués dans
des processus cellulaires tel que la prolifération ou l'invasion qui ont
un rôle important dans la progression tumorale (Fig 15) (Koh et
a!, 2000).

Figure 15: role de la béta caténine (Koh et
al, 2000).
TableauVI : liste des gènes cibles de la â
caténine dans les cancers du colon humain. (Kolligs et a!,
2002).
|
Gènes cibles
|
Direct
|
Activés sous l'effet de la â
caténine
|
|
C_myc
|
oui
|
oui
|
|
Cyclin D1
|
oui
|
oui
|
|
Tcf-1
|
oui
|
oui
|
|
PPAR Delta
|
oui
|
oui
|
|
c-jun
|
oui
|
oui
|
|
Fra-1
|
oui
|
oui
|
|
u-PAR
|
ND
|
ND
|
|
MMP-7
|
oui
|
oui
|
|
Axin-2
|
oui
|
oui
|
|
Nr-CAM
|
oui
|
oui
|
|
ITF-2
|
oui
|
oui
|
2.3. La régulation de l'activité de la
â caténine 2.3.1. Les protéines GSK-3b et Axine
Dans toutes les cellules normales, la
sérine/thréonine GSK3b joue un rôle clé dans le
processus de dégradation de la béta caténine. En
phosphorylant la béta caténine sur plusieurs sites consensus
situés dans son domaine aminoterminal, elle permet une reconnaissance de
la protéine par des ubiquitines ligases. La béta caténine
ubiquitinylée est alors rapidement dégradée par le
protéasome. Plusieurs études ont notamment démontré
que des délétions de la partie aminoterminale de la béta
caténine conduisent à son accumulation dans le cytosol. In
vitro, la béta caténine est peu phosphorylée par
GSK3b car il n'y a pas d'interaction directe entre ces deux protéines.
In vivo, dans le complexe protéique quaternaire : béta
caténine, GSK3b et protéine Apc. Elle contribue au rapprochement
de la ligase GSK3b et ses substrats et facilite leur phosphorylation.
(Ikeda et a!, 1998) ; (Hart et a!, 1998).
Dans ce complexe, la phosphorylation de la protéine Apc augmente son
association avec la béta caténine et la phosphorylation de cette
dernière. (Rubinfield et al, 1996). La phosphorylation
de l'axine/conductine par GSK3b augmenterait la stabilité de la
protéine.
L'accumulation de la béta caténine dans les
tumeurs colorectales résulte essentiellement d'altérations
géniques de l'un de ses partenaires, mais cependant aucune mutation au
niveau de GSK3b n'a été détectée (fig 16),
(Reya, Clevers, 2005).

Figure 16 : Régulation de la â
caténine la GSK-3b (Reya, Clevers,
2005).
La GSK-3b est elle aussi sujette à de nombreux
mécanismes de régulation. Elle peut être inhibée par
phosphorylation en sérine9 (Tsujio et a!, 2000)
; (Grimes et Jope, 2001). De nombreuses kinases ont
été décrites pour être responsable de ces
modifications comme la p 70S6kinase, p 90rsk (leur effet peut être
réversible par la PP2A) (Sutherland et a!,
1993), Akt (Cross et a!, 1995). ILK
(Intergin Linked Kinase) (Delcommenne, 1998), des
protéines kinases (Cook et a!, 1996) ; (Tsunjio et
a!, 2000) et des protéines kinases dépendantes
d'AMP cyclique (Protéine Kinase A) (Li et al, 2000). La
formation de complexes protéiques, médiées par les
protéines associées à la GSK-3b, est également un
autre moyen pour la réguler. En effet, rappelons que
l'association de la GSK-3b au sein du complexe APC/Axine/ â
caténine est responsable de son activation.
La fixation de Frat-1 sur la GSK-3b, facilitée par la
protéine Disheveled, entraine son inhibition et sa séparation de
l'axine (Li et a!, 1999) ; (Tsujo et a!, 2000) ;
(Grimes et Jope, 2001). Enfin la GSK-3b peut être activée
par phosphorylation en tyrosine 216, ce qui augmente son activité
enzymatique. Le sort et son équipe ont montré que Fyn pourrait
directement phosphoryler la GSK-3b en tyrosine 216 et donc l'activer.
(Lesort et a!, 1999).
De plus, une élévation transitoire de la
concentration intracellulaire de Ca2+entraîne également
l'activation de la GSK-3b (Hartigan et Johnson, 1999).
2.3.2. Régulation de la â caténine
par APC
L'Apc se lie à la béta caténine
(Rubinfield et a!, 1993) ; (Su et a!, 1993)
qui a été originellement associé à la zone
d'adhérence des cellules épithéliales. La béta
caténine se lie au domaine cytoplasmique des cadhérines et
à l'alpha caténine. Elle est reliée au réseau
d'actine via l'alpha caténine. L'Apc se lie à la béta
caténine par deux régions différentes (fig 17) (Su
et a!, 1993). Bien que de nombreux mutants de l'Apc aient
perdu au moins l'un des deux sites de fixation, ils gardent la capacité
de se lier à la béta caténine.

Figure 17: interaction entre béta caténine
et Apc (Su et al, 1993).
L'Apc pourrait jouer un rôle important dans
l'adhérence cellulaire par ses capacités de liaison avec la
béta et gama caténine (plakoglobine) en modulant l'expression de
béta caténine libre et fixée dans la cellule. En effet,
certaines lignées de cellule du colon qui expriment l'Apc mutée
contient des taux importants de béta caténine. La transfection de
ces cellules avec l'Apc sauvage ou avec des fragments contenant la partie
centrale des 20 acides aminés réduit significativement la
quantité totale de la béta caténine. (Munemitsu et
a!, 1995). Une surexpression de l'apc sauvage induit une
augmentation du catabolisme de la béta caténine. La région
de l'Apc responsable de la régulation de la béta caténine
est localisée dans la partie centrale de la molécule, qui est
également le domaine phosphorylable par la GSK3b. (Rubinfield et
a!, 1996). La forme phosphorylée de l'Apc aurait une
affinité plus importante pour la béta caténine que la
forme non phosphorylée. L'hypothèse émise est que l'Apc
« pressentirait » l'excès de béta caténine
intracellulaire. Elle s'associerait alors plus fortement à la GSK3b qui
la phosphoryle et à la béta caténine
pour en démarrer la dégradation
protéolytique, via la voie ubiquitine-protéasome. (Aberle
et al, 1997).
2.3.3. La fixation de Disheveled au sein du complexe
multi protéique
Les analyses génétiques de Dsh l'ont
défini comme un médiateur positif de la voie Wnt,
positionné en aval du récepteur et en amont de la â
caténine. (Noordemer et al, 1994). La famille
de protéine Dsh ne possède pas de fonction enzymatique connue,
mais comporte de nombreux sites d'interaction potentiels avec de nombreuses
protéines ; un domaine NT DEP. Il a été décrit chez
la drosophile, que l'expression ectopique de Dsh activait la voie Wnt
(Yanagawa et al, 1995). Disheveled, a trouvé
son homologue chez les mammifères (Dv1), ainsi que sa place au sein de
la voie Wnt. En effet, l'interaction de la protéine Wnt avec Frizzled va
activer la protéine Disheveled selon des mécanismes encore mal
connus. En revanche, il a été montré après
activation de la voie Wnt que DvI et l'axine interagissent par leurs domaines
DIx respectifs (Kishida et al, 1999). La liaison Dv1
sur l'axine déstructure le complexe de dégradation. Les kinases
n'ont donc plus accès à la â caténine qui peut ainsi
s'accumuler dans le cytoplasme. (Sub et al, 2001). De
plus, il a été démontré que la caséine
kinase Ie active ( CKIe) régule positivement la voie de signalisation
Wnt en déstabilisant le complexe de dégradation. Elle agit en
aval de Dv1 mais en amont de la GSK-3b. (Gao et al,
2002). Une 3eme kinase, Par-1, est également décrite
comme régulateur positif de la voie Wnt, notamment en interagissant et
en phosphorylant directement Dv1. (Sun et al, 2001).
Un autre mécanisme, régulant négativement la voie Wnt via
Dv1 a été décrite. En effet la protéine I Dax
(Inhibition of Dv1 Axin complex) interagit avec le domaine PDZ de Dv1 et inhibe
la formation du complexe Dv1/Axine. (Hino et al, 2001).
De même une nouvelle protéine nommée Axam
(A Xin Molecule) entre en compétition avec Dv1 pour se lier à
l'axine, sans empêcher GSK-3b, â caténine ou PP2A de s'y
lier. Ces deux protéines empêchent ainsi Dv1 d'inhiber la
phosphorylation de la â caténine par GSK-3b (Kadoya et
al, 2000). Il se pourrait que le signal Wnt lève
l'inhibition pour Axam et I Dax de la liaison de Dv1 à Axine
(Sun et a/, 2001).
2.3.4. Régulation de la 3 â -
caténine par Frat-1
En absence de stimulation de la voie Wnt, l'axine se lie
à la GSK-3b, â caténine et APC, permettant ainsi à
GSK-3b de phosphoryler la 3 â caténine. Dv1 se fixe au complexe en
interagissant avec l'axine, mais ne parait pas jouer un rôle important en
absence de signal Wnt. Dv1 se fixe également à un peptide
nommé Frat-1 (Frequently Rerrangedin AdvandT Cell Lymphonas I). La voie
Wnt mène à la déstabilisation de la GSK-3b de l'axine avec
l'aide de Dv1 et Frat-1. Ceci suggère que dv1 serait capable de recruter
Frat-1 au niveau du complexe comprenant l'axine. Ainsi, il pourrait agir avec
la GSK-3b après stimulation par Wnt. (Li et a!,
1999). La protéine GBP homologue de Frat-1 chez le
xénope a été décrite pour etre capable d'inhiber la
phosphorylation et la dégradation de la â caténine in vitro
(Salic et a!, 2000). Bien que Frat-1 contribue au
développement du cancer dans un modèle de souris
transgénique, son rôle dans les cancers humains n'est pas bien
documenté.
2.3.5. Autres voies de régulation
La compétition entre les différents partenaires
de la â caténine participe à sa régulation. Par
exemple, la surexpression des cadhérines provoque le recrutement de la
majorité de la â caténine au niveau des jonctions
adhérentes, ceci réduit sa capacité à se complexer
avec les facteurs de transcription Tcf/LEF, et inhibe alors la transcription
méfiée par la â caténine. (Orsulic et
a!, 1999).
L'effet inverse est obtenu en sur exprimant le facteur Tcf
(Simcha et a!, 1998). L'état de
cancérisation pourrait donc dépendre du rapport
cadhérines/Tcf/LEF.
Les interactions â caténine /cadhérines
entrent également en compétition avec le complexe de
dégradation (APC, Axine....) protégeant ainsi la â
caténine, impliquée dans l'adhésion cellulaire, de la
dégradation par le protéasome.
2.4. Mécanismes de dégradations de la
â carénine 2.4.1. Le protéasome
Le complexe formé autour de l'axine mène à
la dégradation de la â carénine par le protéasome
à l'aide de la kinase GSK-3â. En effet, la â caténine
posséde une
séquence consensus DSGXXS dont les
phosphosérines (S33 et S37) sont reconnues par la â-TrCP.
(Kitagawa et a!, 1999). L'ubiquitination des
protéines requière l'action conjuguée de trois enzymes qui
forment une cascade de réactions de transfert d'ubiquitine ligase : une
enzyme d'activation E1, une enzyme de conjugaison E2 et une ubiquitine ligase
E3 (Kitagawa et a!, 1999). Directement après
avoir été phosphorylée par GSK-3â, la â
caténine est prise en charge par â TrCP qui catalyse son
ubiquitination. Le protéasome 26S va alors pouvoir la reconnaître
et la dégrader. Â-TrCP est lui-même un des gènes
cibles de la voie Wnt. Ainsi, plus les gènes cibles de Wnt sont
activés, plus il y aura de â-TrCP pour dégrader la â
caténine. C'est une boucle de rétrocontrôle
négatif.
Des résultats récents montrent que siah-1
(homologue humain du Drosophilia Sevenln Absentia), protéine dont
l'expression est sous le contrôle de P53 et qui est impliqué dans
l'arrêt du cycle cellulaire, a également la capacité de
dégrader la â caténine. Cette dégradation est
indépendante de la phosphorylation de la â caténine par
GSK-3 â. Elle ne nécessite pas la protéine â-TrCP
servant de récepteur à la â caténine
phosphorylée et constituant le premier signal pour l'ubiquination et la
dégradation de la â caténine (Liu et a!,
2001). Cependant, une autre protéine, Eb1 est
utilisée par siah-1 dans le but d'ubiquitiner et de dégrader la
â caténine (Fig18) (Matsuzawa et Reed, 2001).

Figure 18 : dégradation de la béta
caténine par le protéasome
(Matsuzawa et
Reed, 2001).
Conclusion
| 
