|

REPUBLIQUE ALGERIENNE DEMOCRATIQUE ET
POPULAIRE
ET DE LA RECHERCHE SCIENTIFIQUE
UNIVERSITE
MENTOURI-CONSTANTINE
FACULTE DES SCIENCES EXACTES
DEPARTEMENT DE PHYSIQUE
:
Série :
MEMOIRE
Présenté pour obtenir le diplôme
de Magister en Physique
Spécialité : Sciences des
Matériaux
Option : Métallurgie
THEME
Caractérisation de la réaction interfaciale
entre une
Par:
KHECHBA Mourad
Soutenu le : / / 2008
Devant le jury:
Président : A. BOUABELLOU Prof. Université
Mentouri-Constantine
Rapporteur : R. HALIMI Prof. Université
Mentouri-Constantine
Examinateurs : S. CHEKROUD Prof. Université
Mentouri-Constantine
M. BENKERRI M.C. Université Ferhat ABBAS
Sétif
|
Caractérisation de la réaction interfaci
|
|

In this work, we characterize the reaction of tungsten thin
layer (6um thickness) deposited by rf magnetron sputtering technique at
500°C, during 60min, with a steel XC70 substrate (according to AFNOR)
containing 0,7 % of carbon.
The samples (thin layer of tungsten + steel substrate) were
submitted to thermal treatments in vacuum at various temperatures
(500°C-1000°C) and during different times. The specimens are
characterized by X ray diffraction (XRD), optic microscopy, scanning electron
microscopy (SEM) and by Vickers tests for the measurements of
micro-hardness.
The main obtained results can be summarized as follows:



In the temperature range 500-800°C, no formation of
tungsten carbides was observed. However, the annealing at a temperature greater
than or equal to 900°C promotes the reaction between the constituents of
the samples (W, Fe, C), and hence the formation of W2C carbide. No other
compounds were detected.
The micro-hardness measured by Vickers tests, increases with
the rise in temperature, particularly from 900°C. The increase in the
hardness is due, probably, to the formation and growth of W2C carbide.
It is also shown that, the micro-hardness decreases with
increasing of the applied load and the depth of penetration until substrate.
For thin coating (2 and 4 m):
W2C carbide is formed with annealing at 800° C for 30 min
for all samples.
The formation of ternary carbide Fe3W3C takes place after
annealing at T C.
The mono carbide WC is obtained with an annealing at
1000°C for 60 min, only in the case of the thinner coating (2 m).

The morphology of the surface samples depends on the temperature
and duration of thermal annealing.
Key words: Thin films,
magnetron sputtering, coating, tungsten, carbides.
Introduction générale :
L'élaboration de matériaux sous la forme de films
minces présente un intérêt majeur
biomatériaux et les outils de coupe.
Les matériaux élaborés sous la forme de
films minces possèdent parfois des propriétés
physico-chimiques différentes en de nombreux points de celles des
matériaux massifs. La nature chimique, la morphologie et la structure
cristallographique du film sont en effet

à déposer et le substrat, lors de
présenté a été
réalisé.
Les carbures et les nitrures des métaux de transition
sont fréquemment les plus utilisés grâce à leur
réfractérité exceptionnelle et leurs
propriétés remarquables : stabilités thermiques,
duretés plus élevées et faible résistivité
électrique [1-2]. Divers procédés, tels que les
dépôts physique en phase vapeur (PVD) et chimique en phase vapeur
(CVD), permettent de produire ces revêtements sous forme de couches
minces ou multicouches.
Le but de ce travail est la caractérisation de la
réaction interfaciale des couches minces de tungstène
déposées par la technique de pulvérisation cathodique RF
à la température 500°C pendant 60 min, avec des substrats
d'acier d'une nuance XC70 contenant 0,7% en poids de carbone (d'après
AFNOR).

Notre mémoire chapitres :
Dans le premier chapitre, nous présentons, dans une
première partie, les méthodes d'élaboration les plus
couramment utilisées pour la réalisation des couches minces,
chapitre, nous décrivons des définitions simples
sur le phénomène de diffusion, puis leurs mécanismes et
leurs divers types.



Dans le deuxième chapitre, nous présentons des
généralités sur les carbures des métaux de
transition. Deux grandes parties ont été
développées : les carbures des métaux de transition, puis
les carbures de tungstène.
le troisième chapitre, est consacré à les
procédures expérimentales suivies pour réaliser les
couches minces des carbures de tungstène, nous décrivons aussi
dans ce chapitre les différentes méthodes de
caractérisation utilisées telles que: La diffraction des rayons X
(DRX), le microscope optique, la microscopieélectronique à
balayage (MEB), et les essais Vickers pour les mesures de la
micro-dureté.
Les résultats expérimentaux concernant les
proprëtés structurales, morphologiques et mécaniques des
couches minces des carbures de tungséne seront éxposés et
discutés dans le quatrième et dernier chapitre.
importants obtenus.
|
Fichiers ASTM.
|
|
Iron
Iron,syn
|
|
Int
|
h k l
|
|
Rad : CuK 1 : 1,45056 Filter: Ni Beta d-sp :
|
44,673
|
100
|
1 1 0
|
|
Cut off: Int: Diffract. I/Icor:
|
65,021
|
20
|
2 0 0
|
|
Ref: Swanson et al, Nati. But. Stand. (U.S.),
Circ.539,IV,3(1955)
|
82,333
|
30
|
2 1 1
|
|
Sys: Cubic S.G: Im3m (229)
|
98,945
|
10
|
2 2 0
|
|
a: 2,8664 b: c: A: C:
|
116,385
|
12
|
3 1 0
|
|
Ref: Ibid.
|
137,136
|
6
|
2 2 2
|
|
Dx:2,875 Dm: SS/FOM: F6=225(0044,6)
|
|
|
Color: Gray, light gray metallic.
|
|
|
Pattern taken at 25°C. CAS #. 7439-89-6. The iron used was
an exceptionally pure rolled sheet prepared at the NBS Gaithersburg,
|
|
|
MD, USA, [Moore, G, J, Met, 5 1443(1953)] It was annealed in an
H2 atmosphere for 3 days at 1100°C and slowly cooled in an
|
|
|
H2 atmosphere. Total impurities of sample<0, 0013%·each
metal
and non-metal. - -Fe(bcc). Opaque mineral
optical data on specimen
from Meteorite: RR2Re=57,7, Disp=16
|
|
|
VHN= 158 (mean at 100, 200, 300), Color values=311, 316, 57,9,
|
|
|
Ref: IMA Commission on Ore Microscopy QDF. W type. Iron group,
iron subgroup. Also called: ferrite. PSC: c12. Mwt: 55,85.
|
|
|
Volume [CD]:23,55.
|
|
|
W
Tungsten
|
|
Int
|
h k l
|
|
Rad : CuK 1 : 1,4505 Filter : Ni Beta d-sp :
|
40,262
|
100
|
1 1 0
|
|
Cut off: Int: Diffract. I/Icor: 18,00
|
58,272
|
15
|
2 0 0
|
|
Ref: Swanson, Tatge. Natl. Bur, Stand. (U.S.),
Circ.539,I,28(1953)
|
73,192
|
23
|
2 1 1
|
|
Sys: Cubic S.G: Im3m (229)
|
87,017
|
8
|
2 2 0
|
|
a: 3,1648 b: c: A: C:
|
100,642
|
11
|
3 1 0
|
|
Ref: Ibid.
|
114,917
|
4
|
2 2 2
|
|
Dx:19,262 Dm: SS/FOM: F8=108(0093,8)
|
131,168
|
18
|
3 2 1
|
|
Color: Gray metallic.
|
153,571
|
2
|
4 0 0
|
|
Pattern taken at 26°C. Sample prepared at Westinghouse
Electronic
|
|
|
|
|
|
|
|
Corp.CAS #. 7440-33-7. Analysis of sample shows SiO2 0, 04%,
|
|
K0,.5%, Mo, Al2O3 and 0,01% each. Merck Index,
8th, p. 1087. W type. Also cooled: Walfram, PSC: c12, Mwt: 183,85.
Volume
|
|
|
|
|
[CD]:31.
|
|
|
|
|
Fichiers ASTM.
|
|
W2C
Tungsten Carbide
|
2
|
Int
|
h k l
|
|
Rad : CuK 1 : 1,450589 Filter: Graph Mono d-sp :Diff
Cut off:17,7 Int: Diffract. I/Icor:
Ref: Natl. Bur, Stand. (U.S.), Monogr.25,21,I,28 (1984)
|
34,524
|
25
|
1 0 0
|
|
38,029
|
22
|
0 0 2
|
|
39,569
|
100
|
1 0 1
|
|
Sys: Hexagonal S.G: P3ml (164)
a: 2,99704(9) b: c: 4,7279(3) A: C:1,5775
1 mp:
Ref: Ibid.
|
52,300
|
17
|
1 0 2
|
|
61,861
|
14
|
1 1 0
|
|
Dx:17,144 Dm: SS/FOM: F22=93(0085,28)
|
69,769
|
14
|
1 0 3
|
|
Color: Dark gray.
Peak height intensity. The mean temperature of data collection
was 23,5°C. The sample was obtained from CEKAC,
Incorporated. Milwaukee, Wi, USA, CAS #. 12070-13-2.
obs)=#177;0,01. Cd 12 type. Tungsten
used as an internal stand. PSC: hP3. To remplace 2-1134. Mwt: 379,71. Volume
[CD]: 36,78.
|
72,839
|
2
|
2 0 0
|
|
74,979
|
12
|
1 1 2
|
|
75,984
|
10
|
2 0 1
|
|
81,328
|
2
|
0 0 4
|
|
85,228
|
3
|
2 0 2
|
|
91,472
|
2
|
1 0 4
|
|
100,510
|
3
|
2 0 3
|
|
103,472
|
<1
|
2 1 0
|
|
106,614
|
5
|
2 1 1
|
|
112,189
|
3
|
1 1 4
|
|
116,452
|
1
|
2 1 2
|
|
120,239
|
2
|
1 0 5
|
|
123,650
|
1
|
2 0 4
|
|
125,841
|
1
|
3 0 0
|
|
135,299
|
3
|
2 1 3
|
|
142,932
|
2
|
3 0 2
|
|
WC
Tungsten Carbide
Unnamed mineral, syn [NR]
|
|
Int
|
h k l
|
|
Rad : CuK 1 : 1,45056 Filter: Mono d-sp :
Cut off: Int: Diffract. I/Icor:
Ref: Bind. MeCarthy, G, Penn State, University Park, Pa, USA,
ICDD, Grant-in-Aid,(1973).
|
31,474
|
45
|
0 0 1
|
|
35,626
|
100
|
1 0 0
|
|
48,266
|
100
|
1 0 1
|
|
Sys: Hexagonal S.G: P6m2(187)
a: 2,9062 b: c: 2,8378 A: C:0,9765
Ref: Ibid.
|
63,979
|
20
|
1 1 0
|
|
65,701
|
6
|
0 0 2
|
|
Dx:15,669 Dm: SS/FOM: F14=29(0035,14)
|
73,064
|
25
|
1 1 1
|
|
Sample obtained from General Electric, type KB, lot no.
131412. Described as a mineral from Mengyin, Shadong and Danba, Sichuan, China.
Zianhong, Z, Guojie, Y, Zhaohni, L, Acta Mineral. Sinica, 6 344-349 (1986). CAS
#. 12070-12-1. CW type. PSC : hp2.To replace 5-728. Mwt :195-86. Volume [CD]
:20,76.
|
75,442
|
14
|
2 0 0
|
|
77,101
|
30
|
1 0 2
|
|
84,015
|
20
|
2 0 1
|
|
98,734
|
14
|
1 1 2
|
|
108,186
|
10
|
2 1 0
|
|
109,026
|
1
|
0 0 3
|
|
109,712
|
10
|
2 0 2
|
|
117,292
|
20
|
2 1 1
|
|
|
|
Fichiers ASTM.
|
|
Fe3W3C
Iron Tungsten Carbide
|
|
|
Int
|
h k l
|
|
Rad : CuK 1 : 1,4518 Filter : Mono d-sp :Diff
Cut off: Int: Diffract. I/Icor:
Ref: Azubike,D.Chrysunthou, A. Lgiabon, U.Private,
Communication, 7, 162, (1992).
|
13,759
|
5
|
1 1 1
|
|
15,942
|
<3
|
2 0 0
|
|
26,590
|
<3
|
3 1 1
|
|
Sys: Cubic S.G: Fd3m(227)
a: 11,1094 b: c: A:
Z: 16
Ref: Ibid.
|
C: mp:
|
32,203
|
22
|
4 0 0
|
|
35,183
|
30
|
3 3 1
|
|
Dx:14,167 Dm: SS/FOM: F25=183(0037,37)
|
39,714
|
50
|
4 2 2
|
|
Equilibrated in evacuated silica capsules at 1200°C for 5
days. C Fe3W3 type.
~ not permitted by space group
PSC: cF112. To remplace 3-980. Mwt: 731.10. Volume
[CD] :13,71.11.
|
42,235
|
100
|
5 1 1
|
|
46,185
|
36
|
4 4 0
|
|
49,166
|
4
|
4 4 2
|
|
52,017
|
4
|
6 2 0
|
|
54,764
|
4
|
6 2 2
|
|
59,361
|
16
|
5 5 1
|
|
64,361
|
16
|
5 5 3
|
|
67,377
|
4
|
8 0 0
|
|
69,159
|
20
|
7 3 3
|
|
72,075
|
52
|
8 2 2
|
|
73,807
|
18
|
5 5 5
|
|
78,350
|
7
|
9 1 1
|
|
78,905
|
5
|
8 4 2
|
|
81,145
|
5
|
6 6 4
|
|
87,294
|
22
|
7 7 1
|
|
89,994
|
13
|
10 2 0
|
|
91,649
|
16
|
9 5 1
|
|
92,202
|
10
|
10 2 2
|
|
96,063
|
16
|
9 5 3
|
|
98,839
|
10
|
10 4 2
|
1997JCPDS-International Centre for Diffraction Data. All rights
reserved PCPDFWIN v.1.30.
|
Généralité et contexte
bibliographique.
|
mince, et enfin le phénomène de diffusion, leurs
mécanismes et leurs divers type.
Dans ce chapitre, nous présentons des définitions
simples sur les couches minces et
leurs applications, puis nous
décrivons les méthodes d'élaboration les plus
couramment
ouche
I.1. Définition des couches minces:

bidimensionnelle) entraîne une perturbation de la
majorité des propriétés physiques. La différence
essentielle entre le matériau à l'état massif et celle en
couches minces est liée au fait que dans l'état massif on
néglige généralement avec raison le rôle des limites
dans les propriétés, tandis que dans une couche mince ce sont au
contraire les effets liés aux surfaces limites qui sont
prépondérants. Il est assez évident que plus
l'épaisseur sera faible et plus cet effet de bidimensionnelle sera
important, et qu'inversement lorsque l'épaisseur d'une couche mince
dépassera un certain seuil; l'effet d'épaisseur deviendra minime
et le matériau retrouvera les propriétés bien connues du
matériau massif.
matériaux en rapport avec les propriétés
physiques et de la simplicité des technologies mise
couches minces. Citons : les métaux, les alliages, les
composés réfractaires (oxydes, nitrures, carbures), les
composés intermétalliques et les polymères [3].
La seconde caractéristique essentielle d'une couche
mince est que, quelle que soit la procédure employée pour sa
fabrication, une couche mince est toujours solidaire d'un support sur lequel
elle est construite (même s'il arrive parfois que l'on sépare le
film mince dudit support). En conséquence, il sera impératif de
tenir compte de ce fait majeur dans la conception, à savoir que le
support influence très fortement les propriétés
structurales de la couche qui y est déposée. Ainsi, une couche
mince d'un même matériau, de même épaisseur, pourra
avoir des propriétés physiques sensiblement différentes
selon qu'elle sera déposée sur un substrat isolant amorphe tel
que le verre, ou un substrat monocristallin de silicium par exemple.
|
Généralité et contexte
bibliographique.
|
I.2. Quelques tec :
I.2.1. Généralité :
Le dépôt des couches minces sur les
différents substrats est un étape essentielle dans la
majorité des domaines de la technologie moderne, il existe plusieurs
méthodes pour fabriquer des matériaux (semi-
appel à trois composantes différentes, comme
présenté sur la figure I.1:
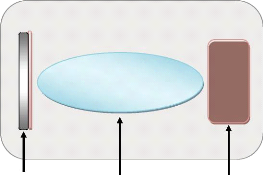
Substrat Milieu Sources du
matériaux pour
dépôt
Figure I.1: Le
procédé du dépôt des couches minces [4].

Une source : le matériau
à déposer est concentré dans un creuset, plaque
métallique

Un substrat : c'est la
pièce à revêtir, c'est ici qu'intervient le
phénomène de condensation.
Un milieu : c'est l'espaces
compris entre la source et le substrat, c'est le siége du
phénomène de transfert de la matière, cela peut être
le siége de réactions chimiques intervenant entre les atomes du
matériau à déposer et un gaz réactif [4].
Le dépôt est formé à partir
d'espèces constituantes (atome, molécules, particules solides, et
particules liquides). Ces espèces viennent d'une source et sont par la
suite transportés vers un substrat à travers un milieu de
type:


Passif
Actif
|
Généralité et contexte
bibliographique.
|
Les principales méthodes utilisées pour
fabriquer des couches minces sous vide font appel à la technique de
dépôt chimique en phase vapeur (CVD : Chemical Vapor Deposition)
[5] et de dépôt physique en phase vapeur (PVD : Physical Vapor
Deposition) [6]. La classification des méthodes est
présentée sur le schéma de la figure I.2 [7].
- Processus Physique
(PVD)

- Processus Chimique
(CVD)
- CVD
-Pulvérisation Cathodique - Processus
thermique
- laser CVD
- plasma CVD

- DC diode - faisceau d'électrons
- DC triode - Implantation d'ions
- RF diode - laser
- RF triode - MBE
Figure I.2: Méthodes
générales de dépôt des couches minces sous vide
[7].
I.2.2. Dépôt chimique en phase vapeur (CVD)
:
Le procédé de dépôt chimique en
phase vapeur consisteà mettre un composé volatil du
matériau à déposer en contact soit avec un autre gaz au
voisinage de la surfaceà recouvrir, soit avec la surface elle-même
(figure I.3). On provoque alors une ou plusieurs réactions chimiques,
donnant au moins un produit solide. Les autres produits de réaction
doivent être
faut systématiquement un apport
énergétique pour favoriser ces réactions. La
réaction est activée par la température du substrat qui
doit être chauffé à une température en rapport avec
le matériau déposé [8].
|
Généralité et contexte
bibliographique.
|
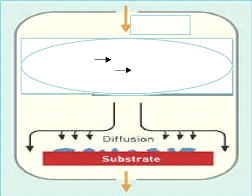
WF gaz+3H2 Wsolide+6HFgaz
WF+H2
Figure I.3: Schéma de
principe de dépôt chimique en phase vapeur (CVD).
A titre d'exemple, le dépôt d'un
film de tungstène très réfractaire peut se faire à
l'aide d'une méthode décrite par l'équation suivante :
600°C
|
WFgaz + 3H2 gaz
|
|
|
Wsolide + 6Hfgaz
|
|
Cette formule implique que si l'on mélange deux gaz WF et
H2, une couche mince de tungstène peut être obtenue. Avec la
méthode CVD, il est possible de déposer des matériaux
La réaction chimique peut être également
activée à l'aide d'un plasma. Cette méthode s'appelle "CVD
plasma" ou PECVD (Plasma Enhanced Chemical Vapor Deposition). Dans ce cas, il y
a création en plus des particulesénergétiques [9].
La fabrication des couches minces métalliques
grâce à ces méthodes s'est particulièrement
développée ces dernières années. Les avantages de
ce procédé sont les suivants : il est facile d'obtenir un assez
grand nombre d'éléments ou de composés chimiques. On
obtient une bonne qualité des couches, un excellent recouvrement des
marches, et une bonne adaptabilité dans une chaîne de production.
En plus, elle offre la possibilité de réaliser des
dépôts sélectifs [5], ce qui permet d'éliminer une
étape de gravure et de planarisation de la surface. Les
inconvénients sont les suivants : les films sont peu denses, ils sont
souvent contaminés par des gaz très réactifs issus de la
réaction chimique (hydrogène, fluor, chlore...), tous les
matériaux ne peuvent être déposés par CVD, et le
système de dépôt est une mise en

|
Généralité et contexte
bibliographique.
|
I.2.3. Dépôt physique en phase vapeur (PVD)
:

Le développement de la technologie de vide a
progressé considérablement pendant les trois dernières
décennies et cela a permis le développement des techniques de
dépôt sous vide telles que les techniques de base de la PVD sont :
l'évaporation et la pulvérisation sous toutes ses formes. Ces
techniques de dépôt permettent l'obtention d'une large
varëté de matériaux à température de
dépôt relativement basse (< 500 °C), par exemple [10].
Films d'oxydes (ZrO2, SiO2, Al2O3, TiO2
Films de nitrures, de siliciures, de carbures, de borures et de
fluorures.
Ces techniques ont été largement utilisées
pour l'obtention des revêtements réfractaires résistants
à l'érosion età l'usure, comme par exemple WC [11-12].
Le dépôt physique en phase vapeur (PVD)
présente beaucoup d'avantages par rapport au dépôt chimique
en phase vapeur, par exemple les films sont denses, le processus est
facileà contrôler et il n'y a pas de pollution [13].
I.2.3.1. Pulvérisation cathodique :
Le phénomène de pulvérisation a
été découvert en 1852 par Grove [14]. Dans un tube
à
utilisation intensive de la pulvérisation comme
moyen de production de couches minces date seulement des
années 50.

La pulvérisation repose sur un principe simple
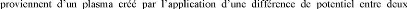
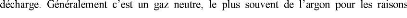
sur lequel croit une couche mince. Dans la technique de
pulvérisation cathodique, les ions électrodes. Le gaz
utilisé pour la création du plasma est appelé gaz
plasmagène ou gaz de suivantes :
(facilité

(le
collision est maximal si les masses des atomes sont proches).
Très faible réactivité chimique.

Faible prix.
|
Généralité et contexte
bibliographique.
|
En pulvérisation cathodique nous distinguons la
pulvérisation simple et la pulvérisation réactive. Dans la
pulvérisation simple l'atmosphère de la décharge est
chimiquement neutre, c'est-à-dire que l'on produit un vide de
10-6 torr. On injecte ensuite le gaz d'argon pur pour créer
le plasma. Dans le cas de la pulvérisation cathodique réactive
l'atmosphère du plasma est réactive, c'est-à-dire que l'on
introduit un certain pourcentage de gaz actif dans l'argon, par exemple de
l'oxygène O2 ou de l'azote N2. Dans chacun de ces cas, la cible peut
être constituée d'un élément simple ou bien d'un
composé. Il existe différents types de systèmes de
pulvérisation cathodique, suivant le mode de création du plasma
ou la nature de la cible (conductrice ou isolante) : diode à courant
continu, triode à courant continu, ou haute fréquence.
a) Principe de la pulvérisation cathodique
DC :
La cible fixée sur une électrode (la cathode) est
portée à une tension négative. Le
à
quelques centimètres (figure. I.4). Après avoir
fa
pression suffisamment élevée (de 10-1
à 1 Pa), une différence de potentiel (de 0,1 à 10
KV)
est appliquée entre les deux électrodes. Une
décharge électrique se produit alors et crée un
tifs du
plasma, attirés par la tension négative de la cathode,
viennent bombarder la cible
ils vont éjecter des atomes de
la cible qui viennent se déposer sur le substrat, formant
ainsi une couche mince. relativement simple et permet de déposer
pratiquement tous les matériaux conducteurs [15].
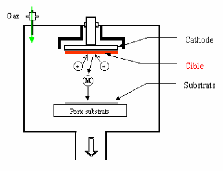
Figure I.4 : Principe de la
pulvérisation cathodique DC .


|
Généralité et contexte
bibliographique.
|
b) Principe de la pulvérisation cathodique
RF :

Les méthodes utilisant une tension continue sont
limitées dans le cas de matériaux
qui arrête le processus de pulvérisati une tension
radio-
alors que penda
la différence de masse et donc de mobilité entre
les électrons et les ions, les électrons étant
beaucoup
plus mobiles. On assiste donc à une accumulation de charges
électroniques à la
cathode. Les courants électroniques
et ioniques vont pouvoir se compenser grâce à
0
isation de la cible.
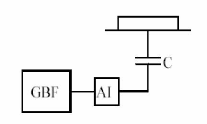
En effet, du
point de vue électronique, le plasma, la cathode et la
cible se comportent comme un circuit

Figure I.5 :
réquence.
c) Pulvérisation cathodique à effet
magnétron :
La décharge électrique continue (DC
and on place derrière la cible des aimants de la
manière présentée dans la figure I.6, on obtient devant la
cible une zone ou le champ magnétique est pratiquement parallèle
à la cible, c'est-à-dire perpendiculaire au champ
électrique [16], et la combinaison des deux oblige lesélectrons
à suivre un trajet non linéaire,
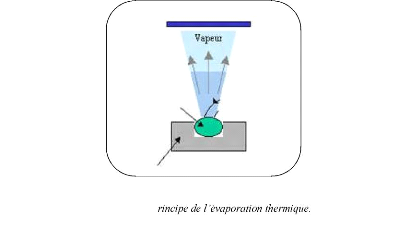
Figure I.7 : P
Cette méthode est la méthode la plus simple car il
n'est pas nécessaire d'injecter un gaz pour créer un plasma,
alors que les autres méthodes PVD ont besoin du plasma comme
Creuset
Matériau
à déposer
Substrat
|
Généralité et contexte
bibliographique.
|
généralement spiridal ou cycloïdal, ce qui
provoque beaucoup plus de collisions avec , 17]. Le seul inconvénient de
cette pulvérisation
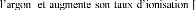
-uniforme de la cible [18].
Figure I.6 : Principe de la
cathode à effet magnétron.
I.2.3.2. Evaporation thermique :
L'évaporation est une technique d'obtention des films sous
vide qui consisteà évaporer ou sublimer un matériau
(figure I.7). Le dépôt se fait par condensation de la phase vapeur
sur
source [19
chauffage électri
champ magnétique, par bombardementélectronique et
par laser [19].
|
Généralité et contexte
bibliographique.
|
intermédiaire. Cependant, certains problèmes
spécifiques à l'évaporation existent: il est difficile de
déposer des matériaux très réfractaires ou à
faible tension de vapeur. Cette méthode ne permet pas de maîtriser
facilement la composition chimique dans le cas d'un alliage par suite d'un
effet de distillation du composant le plus volatil. Les couches peuvent
être aussi contaminées par réaction avec le creuset, avec
le filament et surtout par le dégazage des parois induit par
l'échauffement ou le bombardement desélectrons.
L'évaporation permet l'obtention de film à une vitesse de
dépôt élevée. En revanche, les inconvénients
que présente cette technique sont l"élaboration de film souvent
sous-
des couches et la nécessité d'une densité de
puissance assez importante pour produire la phase gazeuse des matériaux
ayant un point de fusion très élevé.
I.2.3.3. Ablation laser:
un faisceau laser impulsionnel. Le faisceau est
focalisé sur une cible placée dans une enceinte sous ultra-vide
(figure I.8). Les impulsions lasers permettent la vaporisation de
matériaux sous forme de plasma. Le panache de matière ainsi
éjectée perpendiculairement à la cible vient se condenser
sur un substrat placé en vis-à-vis pour former un
revêtement [20-22].
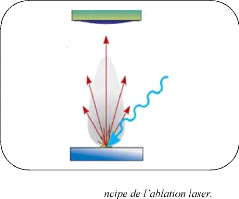
Panacheplasmade
matièreéjectée
Figure I.8 : Pri
Filmmince
Substrat
Cible
Faisceaulaser
Cette technique, connue depuis plus de 20 ans, a prouvé
toute son efficacité concernant
multitude de composés de haute pureté allant
des supraconducteurs à haute température aux
matériaux durs. La pureté des dépôts ne
|
Généralité et contexte
bibliographique.
|
dépend, dans ce cas, que de la pureté de la
cible utilisée. Le principal avantage de cette technique est le
dépôt à température ambiante permettant ainsi le
revêtement sur tout type de substrats allant des semi-conducteurs aux
matériaux polymères [23].
Les lasers utilisés délivrent
généralement des impulsions courtes de durée nanoseconde
(10-9s) ou ultra- uelques centaines de femtosecondes
(10-15s, parfois qualifiées de sub-picosecondes).
I.3. Mécanisme de formation d'une couche
mince:
On peut résumer le processus de croissance d'une couche
mince en disant que c'est une suite statistique de nucléation, puis une
croissance par diffusion de surface et formation d'Ilots, puis une formation
d'Iles de plus grandes dimensions, et enfin la formation d'une couche continue
par remplissage de espèces entre ces Iles. Selon les paramètres
thermodynamiques du dépôt et de la surface du substrat, les
étapes de nucléation et de croissance d'Ilots peuvent être
décrites comme étant [24] :

Du type Iles (appelé Volmer-Weber).
Du type couche (appelé Frank-van der Merwe).
Du type mixte (appelé Stranski-Krastanov).
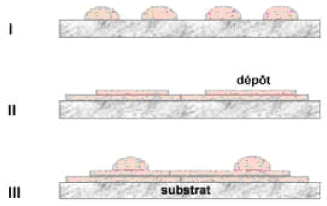
Figure I.9 : Les trois modes de
croissance d'une couche mince, I) Volmer-Weber,
II) Frank-van der Merwe, III) Stranski-Krastanov
[25].
|
Généralité et contexte
bibliographique.
|
La figure I.9 présente les trois types de formation
d'une couche mince. Dans la quasitotalité des cas pratiques, la
croissance de la couche se fait par formation d'îlots, puis d'îles,
puis d'une surface continue. Sauf dans les cas des conditions spéciales
de dépôts (température du substrat, nature et
énergie des espèces incidentes, nature chimique du substrat,
caractéristique
topographiques des îlots sont distribués d'une
façon aléatoire. Ceci signifie que, lorsque ces îlots vont
se rencontrer au cours du processus de croissance, des joints de grains et des
défauts (dislocations diverses ) vont être inclus dans la couche
à la suite des désaccords de configuration
géométrique et d'orientation cristallographique [26].
I.4. Propriétés spécifiques aux
couches minces:
Les propriétés de la matière en couches
minces peuventêtre intrinsèquement différentes. Cela peut
être lié à la microstructure du matériau, à
à la géométrie elle même du compose (effets de
taille) [27] :


Effets des contraintes : pour
les couches en heter
suffisamment fine, le film est contraint (de part la
différence de paramètre de maille entre
à la structure cristallographique. Ce
couplage entre la structure et les propriétés
physiques est important.
effets de taille : par le
terme (effets de taille), on entend les effets physiques liesà la
géométrie du matériau. Dans des structures avec au moins
une dimension inférieure a une longueur caractéristique (comme le
libre parcours moyen desélectrons, la distance tunnel ou les longueurs
de corrélation magnétique), on observe des effets originaux. Par
exemple, les effets de confinem
électronique, modifient radicalement les niveaux
d'énergie et les propriétés radiatives
du
matériau. On peut aussi citer le cas des phénomènes
de transport dépendant du spin. Ils
inférieure au libre parcours moyen électronique.
Schématiquement, cela revient à dire
magnétique.
|
Généralité et contexte
bibliographique.
|
: souvent
différente de la structure dans le massif. La modification
de la coordination des atomes, la présence de liaisons pendantes, ainsi
que les interactions électrostatiques entre le
I.5. Phénomène de diffusion:
Le phénomène de diffusion est un
phénomène très général dans la nature, qui
correspond à la tendance à l'étalement d'espèces
particules, atomes ou molécules grâce à une excitation
énergétique apportée par la chaleur. Suivant le milieu
dans lequel se déplacent ces espèces, l'étalement sera
plus ou moins grand [28].
A température ambiante, le phénomène de
diffusion sera très important dans un milieu gazeux, plus faible dans un
milieu liquide et pratiquement nul dans un milieu solide. Pour obtenir un
phénomène de diffusion dans un solide ou un cristal, il faudra
chauffer le matériau à des températures voisines de 1000
°C
Dans la suite, nous nous intéresserons essentiellement aux
mécanismes de diffusion dans le milieu solide.
I.5.1. Les mécanismes de diffusion atomique
:
La diffusion est la migration d'une espèce sous le seul
effet de l'agitation thermique. La migration peut aussi avoir lieu sous l'effet
d'une force, par exemple une forceélectrostatique dans le cas
d'espèces chargées, ou bien une force chimique; dans ce cas, la
diffusion se superpose et se combine à cette migration [28].
Les mécanismes de diffusion mis en jeu dans un cristal
vont dépendre de la nature du cristal et de la nature de l'espèce
diffusante. On peut noter que les atomes constituant le cristal peuvent eux-
est importante puisqu'elle permet notamment de créer des
lacunes dans le réseau cristallin.
Les mécanismes ne seront possibles qu'à la
condition que les espèces concernées soient suffisamment
excitées (niveau d'énergie suffisant) pour statistiquement
franchir les barrëres d'énergie que constituent par exemple les
barrëres de potentiel atomique. Ces barrëres de potentiel vont
dépendre des mécanismes mis en jeu. Quatre principaux types de
mécanisme de diffusion atomique peuvent exister [29].
|
Généralité et contexte
bibliographique.
|
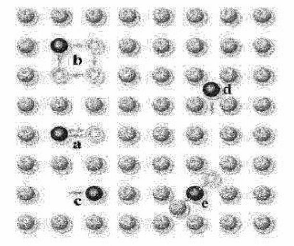
Figure I.10 : Mécanismes
de diffusion dans un solide monoatomique : (a) Echange direct,
(b) Echange cyclique, (c) Mécanisme
lacunaire, (d) Mécanisme interstitiel direct,
(e) Mécanisme interstitiel indirect.

an
décrivant un anneau (cas b dans la figure I.10).



Les mécanismes
interstitiels Les atomes peuvent occuper un site interstitiel
sans induire une déformation importante du réseau
propre de réseau (cas d et e dans figure I.10).

Le mécanisme lacunaire
correspondant à l'occupation d'une lacune ou vacance laissée par
un atome du réseau cristallin qui peut, lui aussi se mouvoir en laissant
des lacunes [28 iculièrement dans les métaux (cas c dans figure
I.10) [29].
|
Généralité et contexte
bibliographique.
|
I.5.2. Les équations de diffusion:
I.5.2.1. Première loi de Fick:
Considérons un élément (A)
dissout à l'intérieur d'une matrice cristalline. A basse
température le système est figé et l'impureté est
immobile. Par contre, Si la température est suffisamment
élevée, sous l'effet de l'agitation thermique, les atomes deA
sont mobiles et ils peuvent se déplacer à l'intérieur
de la matrice. Ils sont alors soumis au phénomène de diffusion :
une non-uniformité des variables thermodynamiques (une variation de la
concentration de (A), toutes choses égales par ailleurs, en
étant le cas le plus fréquent) entraîne un transport de
matière (un déplacement des atomes (A)) à
l'intérieur du solide. La diffusion est donc un processus non
réversible dont le "moteur" est le gradient de potentiel chimique et qui
a pour objectif d'uniformiser ce potentiel chimique. La description
phénoménologique générale de ce processus n'est
possible qu'à l'aide de la théorie de la thermodynamique des
processus irréversibles [30].
Cependant, dans le cas le plus simple d'une seule
espèce diffusante soumise uniquement à son gradient de
concentration, le phénomène se décrit très
simplement à l'aide de la première loi de Fick, qui stipule que
le flux d'impureté J (nombre d'atomes traversant l'unité
de surface par unité de temps) est proportionnel au gradient de
concentration C de l'impureté [30] :

j D C . (I.1)
Le signe "-" de l'équation précédente
traduit simplement le fait que la diffusion a lieu des zones à fortes
concentrations vers les zones à faibles concentrations, ce qui est
naturel pour pouvoir homogénéiser les concentrations.
Par définition, le coefficient de
proportionnalité D est le coefficient de diffusion. Le flux
étant généralement exprimé en atomes par
(cm-2s4) et les concentrations en nombre
d'atomes par centimètre cube (cm3), D
s'exprime alors en cm2s1.
D dépend fortement de la température;
quasi-nul à la température ambiante, ce coefficient est de
l'ordre de l'unité en micron carré par heure pour des
températures de l'ordre de 1100 °C.

17
Généralité et contexte
bibliographique.
C
t
I.5.2.2. Deuxième loi de Fick:
En régime non permanent, le flux de diffusion et la
concentration sont fonction du temps et de la position. Afin de pouvoir
déterminer le coefficient de diffusion, il est nécessaire de
prendre en compte uneéquation de conservation de la matière. Dans
le cas où les particules ne participeraient pas à une
réaction (réaction chimique, réaction entre les
différents types de
[30,31] :
En combinant les équations (I.1) et (I.2), on obtient la
deuxième loi de Fick:
Dans le cas de cristaux cubiques ou de systèmes isotropes,
le tenseur de diffusion se
réduit à un scalaire D
j DgradC
Si de plus la concentration ne varie que dans la
directionx, .3) se réduit à:
C
D
C
t x
x
.. . (I.4)
Si enfin, on peut supposer que le coefficient de diffusion est
indépendant de la concentration,
suivante :
Du point
second ordre. Pour la résoudre, il est
nécessaire de définir les conditions initiales et les conditions
aux limites [32]. Ces conditions sont généralement
imposées d'un point de vue expérimental afin de pouvoir
déterminer les valeurs des coefficients de diffusion par comparaison
entre les profils mesurés et ces lois simples. Nous donnerons en-dessous
des solutions pour deux cas pratiques [30-31].
Diffusion depuis une couche mince:
Il s'agit du cas pratique où l'espèce diffusante
est déposée à l'instant initial sous forme d'un film mince
à la surface (x = 0) d'un échantillon très
épais. S'il n'y a pas d'évaporation de l'impureté,
dès que la profondeur de pénétration est très
grande devant l'épaisseur du film initial le profil de concentration
suit une loi gaussienneà l'intérieur de l'échantillon :
C
0
j
t
grad
. (I.2)
2
C
.. . (I.5)
C
t
D
x
2
grad DgradC
.
. . . (I.3)
|
Généralité et contexte
bibliographique.
|
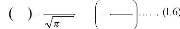
2
x
4 Dt
C x ,
t
Q
Dt
exp
Où Q est la quantité totale
d'impureté déposée (et donc dissoute dans le
matériau) :
0

Q
Cx
dx

Diffusion avec une concentration superficielle constante
:
Il s'agit du cas où une source extérieure
infinie permet de maintenirà la surface de l'échantillon une
concentration constanteC0 (constante par rapport au temps).
La solution fait alors appel à une fonction classique des
statistiques, la fonction erreur complémentaire:
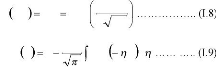
C x t C erfc
, 0
x
2 Dt
u
2 2
1 exp d .
0
erfc
u
Des tables donnent les valeurs de la fonction erreur
complémentaire. Sa variation en
x
fonction de u est donnée sur la figure I.11, en
échelle linéaire et semi-
2 Dt
logarithmique.
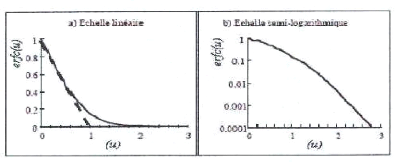
Figure I.11: Fonction erreur
complémentaire.

19
I.5.3. Expression du coefficient de diffusion en
fonction de la température :
. Elle contrôle la
concentration et la mobilité des défauts et par
ce biais la mobilité des atomes au sein du réseau. Dans un
cristal, chaque atome oscille autour de son site qui représente sa
position
amplifient et augmentent ainsi les
identique pour tout autre mécanisme), on peut montrer que
la fréquence de sauts atome
multipliée par la fréquence moyenne
x
Où N: est la fraction atomique de Boltzmann
[32].
N exp
kT
G f
Où f formation des lacunes (en eV), k la
constante de
Boltzmann et T la température (en Kelvin).
Pour sauter
caractéristique de la structure du cristal et de la nature
des proches voisins. Pour cela, il faut
fournir une certaine énergie enthalpie libre de migration
m fait
au La probabilité que
m nécessaire à franchir la barrière
est
donnée par:
kT
Gm
p exp
.... (I.12)
On peut ainsi exprimer la fréquence moyenne de saut
Et donc la fréquence de saut :
exp
kT
Gm
|
Généralité et contexte
bibliographique.
|
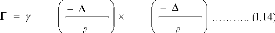
Gf
kT
Gm
kT
exp
exp
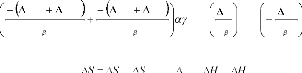
Le coefficient de diffusionD étant proportionnel
à la fréquence de saut longueur du saut [32], on
peutécrire :
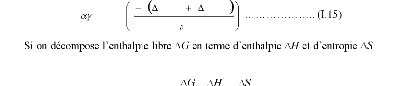
D exp
la relation thermodynamique :
Gm
Gf
kT
= T
suivant
on obtient :
D
Sm

k
S f
exp
S
k
. exp
kT
H
exp
Hm
kT
H f
avec: f + m et H = f + m
On peut écrire, ainsi, le coefficient de
diffusionD, quel que soit le mécanisme de diffusion, de la
façon suivante :
D D0exp

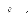
kT
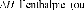
Où D0 est le coefficient préexponentiel
de diffusion (en m2.s-1),
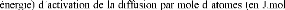
-1) et R la constante des gaz
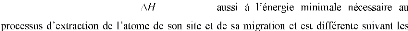
parfaits (= 8.32 J.mol-1.K-1).
correspond
mécanismes de diffusion mis en jeu. On
remarqueégalement que le coefficient de diffusion augmente de
manière exponentielle lorsque que la température augmente.
I.5.4. Les divers types de diffusion:
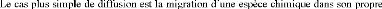
réseau : diffusion de A dans A. Dans ce
cas les sauts sont complètement aléatoires car rien ne
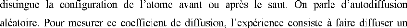
|
Généralité et contexte
bibliographique.
|
atome marqué (par exemple un isotope) dilué dans le
solide pur : diffusion deA* dans A. Ce
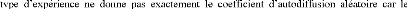
traceur est discernable des atomes normaux du réseau. Le
coefficient sera modifié par un
facteur de corrélation et
aussi par un effet isotopique qui traduit la différence de masse
entre

en position
normale : diffusion deB en faible concentration
dansA.
On désigne par interdiffusion ou diffusion chimique, les
expériences dans lesquelles apparaissent de forts gradients chimiques :
diffusion après mise en contact de deux solides purs A et
B
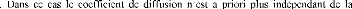
concentration [33].
|
Généralité sur les carbures des
métaux de transition.
|
Dans ce chapitre, nous donnerons uneétude bibliographique
sur les carbures des métaux de transition, et en cas particulier les
carbures de tungstène.
II.1. Carbures des métaux de
transition:
Les carbures sont des combinaisons de carbone avec un autre
élément ou plus, du système périodique, qui peut
appartenir aux groupes IA, IIA, IIIA et peutêtre aussi un métal de
transition du groupe IV, V, VI [34]. Leséléments
métalliques généralement forment des carbures et des
nitrures ; ils peuventêtre divisés en plusieurs types avec
différentes structures et caractéristiques physico-chimiques
[35]. Les carbures et les nitrures des métaux de transition sont des
composés contenant deux types d'atomes dont un type est un métal
de transition, comme le Ti, le W, etc et l'autre est du carbone ou azote. Pour
la brièveté, ils sont marqués comme métal, Me = Ti,
W..., et le non-métal, par Y = C, N, où Me et Y
36].
Les carbures des métaux de transition sont
groupés selon la position du métal de transition dans le tableau
périodique (tableau mondéliéev). Lorsque l'atome de
carbone s'insère dans le réseau, la structure de métal
varie (Tableau II.1).
|
Généralité sur les carbures des
métaux de transition.
|
Tableau II.1 : Les groupes des
carbures des m étaux de transition [37-39].
|
Noms
|
Synonymes
formules
|
Struct. Cristal.
Couleurs
|
Utilisations
|
|
Carbone
|
C
|
amorphe
noir
|
nucléaire
|
|
Carbure de bore
|
B4C
|
rhomboédrique
noir
|
mécanique, nucléaire
|
|
Carbure
|
HfC
|
/
|
carbure
|
|
Carbure de
Niobium
|
NbC
|
Cubique
|
/
|
|
Carbure de
silicium
|
SiC
|
hexagonale ou
cubique
incolore - noir
|
thermique, thermomécanique,
mécanique,
électrique,
électronique, nucléaire, chimique
et
militaire.
|
|
Carbure de
Tantale
|
TaC
|
cubique
noir
|
|
|
Carbure de
Tungstène
|
W2C
WC
|
hexagonale
hexagonale
noir
|
mécanique
|
|
Carbure de titane
|
TiC
|
Cubique
|
mécanique
|
|
Carbure de titane
|
Ti2C
|
Hexagonale
|
/
|
|
Carbure de
Vanadium
|
VC
|
Cubique
|
/
|
|
Carbure de
Vanadium
|
V2C
|
Hexagonale
|
/
|
|
Carbure de Zirconium
|
ZrC
|
Cubique
|
|
|
Graphite
|
C
|
hexagonale
noir
|
fibres réfractaires
|
|
Carbure de
molybdène
|
MoC
Mo2C
|
hexagonale
hexagonale
|
/
|
|
Généralité sur les carbures des
métaux de transition.
|
II.2. Propriétés des carbures des
métaux de transition:
Les carbures des métaux de transition ont une
combinaison unique des propriétés par exemple, un point de fusion
élevé, une haute stabilité chimique, une grande
dureté, une très bonne conductivité thermique et
électrique, et une bonne ésistance à la corrosion [40].
Mais ils sont très fragiles. Toutes ces propriétés
justifient leurs applications technologiques, soit comme matériaux
massifs ou comme des couches minces [41].
II.2.1. Propriétés structurale : a)-
La structure cristaiine :
La plupart des carbures des métaux de transition sont
des composants interstitiels où les atomes de carbone s'incorporent dans
le éseau du métal de transition. La géométrie des
sites interstitiels et la liaison entre le carbone et le métal sont des
caractéristiques nécessaires pour déterminer la structure
cristalline des carbures. La structure des carbures des métaux de
transition se détermine selon la règle de Hägg. Cette
règle est développée empiriquement les structures des
borures, des carbures, des halogénures, et des nitrures en métal
de transition [42].

Pour les composés des métaux de
transition (carbures, nitrur :
Si le rapport r=rX/rM (rX est le rayon
et
rM est le rayon
type B1(NaCl) ou hexagonale oit les atomes
de
atomique du m étal) est inférieur
à 0,59, la structure sera simple de
Règle Je Hägg
non métal occupent les sites
interstitiels octaédriques. C'est pourquoi ces
compos és sont connus par les composés
interstitiels.
Si r est supérieur à 0,59,
l'arrangement m étaiique est distordu pour accommoder les grands atomes
du non m étal et pour préserver l'interaction m
étalmétal. Ilen résulte la formation des structures
complexe.
Selon la règle de Hägg, si le rapport r est
inférieur à 0.59, les atomes de carbone s'insèrent dans
les positions interstitielles, sans provoquer de distorsions du éseau du
métal. Deux types des carbures interstitiels distinguent selon le type
de éseau cristallin des atomes de métal [43].
|
Généralité sur les carbures des
métaux de transition.
|
Dans le première type les carbures sont interstitiels
avec un réseau du métal à symétrie cubique à
faces centrées par exemple (TiC, ZrC, VC, HfC, TaC), dans ce cas et dans
les carbures e s'incorporent dans les sites interstitiels octaédrique ou
dans le centre du prisme trigonal. Par contre, dans le deuxième type les
carbures sont interstitiels avec un réseau du métal à
symétrie hexagonale compacte (par exemple : V2C, Ta2C, Mo2C, W2C), dans
le cas des carbures carbone occupent la moit des sites interstitiels
octaédriques disponibles.
Dans les deux types de carbures interstitiels, la coordinence du
carbone est de 6, età
comme carbone libre mélange au carbure.
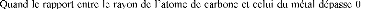
,59
deux autres types fondamentaux de structures se forment
[43-44].
a- Structure avec atomes de carbone isolés ayant une
coordinence de 8 et une symétrie hexagonale ou rhombique (MoC, WC,
Mn3C).
b-
symétrie hexagonale (Cr7C3, Mn7C3).
Généralement, on distingue trois genres de
structure des carbures des métaux de transition (figure II.1) selon le
type de réseau de métal:



La structure B1 (NaCl), où les sites
interstitiels octaédriques de l'élément
métallique,
de type (CFC), sont totalement occupés par les
atomes de carbone. Elle présente le
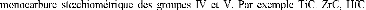
VC, NbC et TaC.
la structure L'3, où les atomes de carbone
occupent la moit des sites interstitiels octaédrique du
sous-réseau de l'élément métallique, de type
hexagonal compact (Hcp s de type M2C. par exemple V2C, Ta2C, Mo2C et W2C.
la structure WC, où les atomes de carbone occupent
les sites interstitiels du centre d'un prisme trigonal du sous réseau de
l'élément métallique, est hexagonal simple.
|
Généralité sur les carbures des
métaux de transition.
|
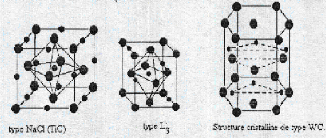
Figure II.1 : Trois genres de
structures des carbures des m étaux de transition.
La règle de Hägg introduit une
géométrie simple pour les structures formées, tant que la
structure des carbures se détermine par leurs propriétés
électroniques. La structure électronique a un effet
considérable sur les changements de la structure cristalline dans les
métaux et les carbures [42].

La progression en structure cristalline d épend du
nombre d'électrons de valence sp par atome (e/a)
:
GLl lliro1ic t'/ngcl-131c r(c1
ée est CC.
la structure formée est Hcp.
ée est CFC.
La caractéristique intéressante de la structure
des carbures des métaux de transition est que le réseau
adopté par le métal dans le carbure n'est jamais celui du
métal de base, c'est-àdire : si le métal a un
réseau Hcp, son carbure a le métal sur un réseau CFC, le
métal de base CFC occupe un réseau non-cubique dans son carbure,
et les métaux de base CC ont des réseaux CFC ou Hcp dans leurs
carbures. Ceci a été expliqué en utilisant la
théorie d'Engel-Brewer [42].
|
Généralité sur les carbures des
métaux de transition.
|
b)- Les liaisons cristallines :
Les composantes réfractaires et parmi elles les carbures
étant connus par la complexité de leurs structures
cristalline.
Il y a trois types [43-45].


La liaison ionique : on trouve ce type de
liaison dans les carbures des métaux alcalins, alcalino-

La liaison covalente : elle est
prépondérante dans les carbures SiC, B4C.
La liaison métallique : les carbures des
métaux de transition présent ce type de liaison.
Dans les carbures covalents, les liaisons sont très
localisées et directionnelles entre les atomes de carbone et ceux de
métal (Si ou B). Il en résulte une haute
réfractivité, une dureté extrême, une
stabilité chimique supérieure à celle de tous
résistance électrique peu élevée
[43,45].
Les carbures métalliques sont caractérisés
par des bondes électroniques délocalisées étendues
sur tous les réseaux
conséquent, quelques propriétés de ces
carbures, comme par exemple la conductivité électrique, sont
spécifiquement métalliques [42].
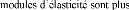
Tous les carbures sont caractérisés par leur grande
dureté et haut point de fusion, les petits pour les carbures ioniques.
Les coefficients de dilatation thermique augmentent en partant des carbur
ioniques [45].
II.2.2. Propriétés physiques et
mécaniques :
Les propriétés physiques et mécaniques
pour certains types des carbures des métaux de transition sont
données dans le tableau suivant (tableau II.2), tout en sachant que ces
propriétés sont très dépendantes de la technique
délaboration. Il existe un grand nombre des composés
possèdent une dureté élevée. Cependant, pour
réussir la conception d'un revêtement, il est nécessaire de
faire une bonne sélection des matériaux ainsi qu'une bonne
sélection de la technique délaboration.
|
Généralité sur les carbures des
métaux de transition.
|
Tableau II.2: Les
propriétés physiques et mécaniques des carbures des m
étaux de transition.
|
matériaux
|
Température
de
fusion
(°C)
|
Dureté
(Kg/mm2)
|
Module
d'Yong
(GPa)
|
Résistivité
électrique
|
Coeff.
moyen
de
dilatation
thermique
|
|
Ti
|
1660
|
55
|
120
|
42,7
|
9,3
|
|
TiC
|
3067
|
3000
|
470
|
50
|
7,4
|
|
TiN
|
2947
|
2000
|
--
|
25
|
9,4
|
|
Zr
|
1855
|
100
|
95
|
42,4
|
7,1
|
|
ZrC
|
3445
|
2800
|
400
|
42
|
6,7
|
|
ZrN
|
2982
|
1600
|
--
|
21
|
7,2
|
|
Hf
|
2223
|
85
|
138
|
32,1
|
5,8
|
|
HfC
|
3928
|
2500
|
352
|
--
|
6,6
|
|
HfN
|
--
|
1600
|
--
|
--
|
6,2
|
|
V
|
1917
|
55
|
130
|
20,1
|
10,0
|
|
VC
|
2830
|
2940
|
430
|
59
|
7,3
|
|
VN
|
2346
|
1520
|
--
|
85
|
9,2
|
|
V2N
|
--
|
1900
|
--
|
--
|
--
|
|
Nb
|
2477
|
80
|
101
|
14,4
|
8,0
|
|
NbC
|
3613
|
1950
|
580
|
19
|
6,6
|
|
NbN
|
2204
|
1420
|
--
|
58
|
10,1
|
|
Nb2N
|
--
|
1700
|
--
|
--
|
--
|
|
Ta
|
3017
|
110
|
186
|
13,1
|
7,0
|
|
TaC
|
3985
|
1700
|
560
|
15
|
6,3
|
|
Ta2C
|
--
|
1300
|
--
|
--
|
--
|
|
Ta2N
|
--
|
1200
|
--
|
--
|
--
|
|
TaN
|
--
|
--
|
--
|
--
|
5
|
|
Cr
|
1857
|
230
|
250
|
12,9
|
10,8
|
|
Cr3C2
|
1895
|
1300
|
390
|
75
|
10,3
|
|
Cr2N
|
1740
|
--
|
--
|
--
|
--
|
|
CrN
|
--
|
1100
|
--
|
64
|
2,3
|
|
Mo
|
2623
|
250
|
320
|
5,47
|
6,0
|
|
Mo2C
|
2520
|
1650
|
230
|
71
|
4,9//a ; 8,2//c
|
|
Mo2N
|
1950
|
1700
|
--
|
79
|
--
|
|
W
|
3422
|
360
|
407
|
5,39
|
4,45
|
|
WC
|
2776
|
2200 (0001)
|
720
|
22
|
5,0//a ; 4,2//c
|
|
|
1300 (1010)
|
--
|
--
|
|
|
WN
|
600
|
--
|
--
|
19,89
|
--
|
|
B4C
|
2450
|
2940
|
450
|
104
|
--
|
|
SiC
|
2300
|
2580
|
480
|
103
|
--
|
|
Si3N4
|
1900
|
1700
|
210
|
1016
|
--
|
|
C (diamant)
|
3800
|
7600
|
7600
|
1018
|
--
|
Les caractéristiques qu'on pourrait tirer du tableau II.2
ci-dessus sont les suivantes : [42, 45,46].
|
Généralité sur les carbures des
métaux de transition.
|
a) - La température de fusion:
Les températures de fusion des carbures et des
nitrures sont très élevées en comparaison avec les
métaux purs, elles sont parmi les plus hautes de tous les types de
matériaux, et elles sont semblables à celles des matériaux
céramiques.
Pour les métaux, le maximum des points de fusion est
observé entre les groupes V et VI, tandis que pour les carbures, il est
dans le groupe V et pour les nitrures dans le groupe IV.
b) - La dureté :
Dans le cas des carbures, la dureté représente
une de leurs proprétés les plus significatives. Les valeurs de la
dureté des carbures sont très élevées en
comparaison avec les nitrures, et les métaux purs, mais ces valeurs de
dureté varient en fonction du mode de préparation du carbure (la
température de préparation, la pression, l'épaisseur de
la couche et la technique de mesure utilisée
Généralement, les métaux les plus durs
ont une énergie de cohésion élevée, une courte
longueur de liaison et une forte liaison de valence. Les carbures des
métaux de transition ayant des liaisons de type ionique,
métallique et covalent. La liaison ionique et métallique
réduit la dureté du cristal.
Dans les couches minces des carbures des métaux de
transition, le renforcement de la
finesse des grains est démontré par la relation de
Hall Petch [47]: H H0 Kd1/ 2
avec:
- H : est la dureté de polycristal.
- H0 : est la dureté
intrinsèque pour un monocristal.
- D : est la taille moyenne des
grains.
- K : est une constante de matériaux.
Mais ils ont remarqué que cette relation est valable pour
les couches monophasées dont la taille de grain est inférieure
à 20 nm.
c) - Le module de Young:
Le module de Young est une autre proprété
mécanique qui représente la rigidité du matériau,
ou bien sa résistance à la déformation
élastique.
Les valeurs de module de Young sont considérablement
grandes pour les carbures et les nitrures comparés à celles des
métaux purs, et elles sont similaires à celles des
matériaux céramiques.
|
Généralité sur les carbures des
métaux de transition.
|
d) - La résistivité électrique
:
Les matériaux peuvent être classifiés comme
conducteurs, semi-conducteurs ou isolants. Les conducteurs
caractérisés par une résistivité dans la gamme de
10-2 3 u
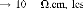
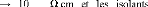
semi-conducteurs approximativement 106 11 u à peu
prés
1013 18u .
Les valeurs de la résistivité électrique
des carbures et des nitrures sont peuélevées que celles des
métaux purs, mais ils sont encore dans la classe de conducteurs. Par
contre, les matériaux céramiques ont des valeurs
extrêmement élevées, donc ils sont des isolants.
e) - La dilatation thermique:
Le coefficient de dilatation thermique (CDT) est une
caractéristique très importante dans l'élaboration des
couches minces. La différence entre les coefficients de dilatation
thermique des substrats et du film est l'origine des contraintes
résiduelles [10]. Les coefficients de dilatation thermique augmentent en
partant des carbures covalents, pui
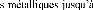
ioniques [45].
La dilatation thermique linéaire peut être
définie en fonction de la température par l'équation
suivante [48] :
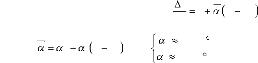
/ C
5 / C 2
10
5
10
0
1
avec : 1 T 25 /
0
1 T 25
l
l
: Le coefficient moyen de dilatation thermique.
En plus de ces propriétés
précédentes, les carbures des métaux de transition sont
caractérisés également par:
f) - La
réfractérité :
Les carbures des métaux de transition des groupes IV
et IIV sont caractérisées par la
réfractérité, c'est-à-dire ils ont un haut point de
fusion (la température de décomposition (tableau II.2)) [49], en
plus ils ont une résistance appropriée aux déformations
mécaniques à une haute température. D'après la
théorie de Branski, la réfractérité des
métaux dépend de la configuration électronique des atomes
de ces métaux [43].
g) - L'adhérence :
L'adhérence de revêtement de carbure avec son
substrat s'exprime comme la force nécessaire pour rompre l'interface.
Plusieurs techniques s'utilisent pour déterminer cette
|
Généralité sur les carbures des
métaux de transition.
|
grandeur, parmi ces tests: le testà rayure (scratch
test). L'adhérence dépend de la température. Elle varie
avec la rugosité de la surface, la dureté du substrat et la
nature chimique de l'interface.
R.Halimi et al. [50] ont étudié
l'adhérence des films minces de carbure de titane en fonction de la
température, ils ont rapporté que l'adhérence augmente
avec la température de recuit jusqu'à 1000 °C et aucune
perte d'adhérence n'a été observée, même pour
une charge de 1000 g.
h) - Les contraintes résiduelles
:

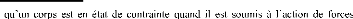
Notion de contraintes :
On dit
extérieures ou de façon plus générale
quand un de ses éléments exerce une force sur les
éléments voisins [51].
Pratiquement toutes les couches mincesélaborées
par PVD présentent des contraintes internes [52]. Ces contraintes
induites par les différentes étapes de dépôt, les
traitements thermiques et thermomécaniques (ou de diffusion) sont dites
contraintes résiduelles [53-54].
Les contraintes résiduelles sont des contraintes
statiques qui existent dans un système isolé soumis à
aucune force ni moment extérieur et qui se trouvent en équilibre
mécanique [55]. Comme les matériaux ne sont pas jamais
parfaitement continus et présentent
Les sources de contraintes peuvent provenir de
déformations aux échelles macro, micro ou submicroscopique.

Ordres de contraintes :
Une classification de contraintes en trois ordres a
été proposée il y a quelques années [51,55].
1- Les contraintes résiduelles du 1er ordre
sont homogènes sur plusieurs grains. Elles peuvent atteindre localement
des valeurs très importantes, mais elles sont nulles à
2- Les contraintes résiduelles du 2eme
ordre
rmations résultantes des désorientations
cristallines entre grains, le retour élastique de chaque grain
étant diffère de celui des autres grains.
|
Généralité sur les carbures des
métaux de transition.
|
3- Les contraintes résiduelles du 3eme ordre
sont homogènes sur les petits domaines de
deux solide (quelques distances interréticulaires). Ces
contraintes sont dues à
1-

2, et 3.
L'origine des contraintes
résiduelles on distingue trois origines de contraintes
dans le revêtement [56] :
résulte de la
Contraintes intrins èques : elles
résultent de la croissance du revêtement. Plusieurs facteurs
engendrent ce type de contrainte, comme le bombardement ionique lors de la
déposition de revêtement qui peut introduire des contraintes de
compression dans la couche superficielle où l'énergie des atomes
qui bombardent est plus grande lorsque la pression est faible. Les
impuretés du gaz introduisent aussi des contraintesélevées
dans les couches minces, la formation d'oxydes aux joints de grains introduits
aussi des contraints de compression. La coalescence des grains et les
transformations de phases influent également sur les contraintes
intrinsèques.
2- Contraintes thermiques : elles
résultent de la différence des coefficients de dilatation
(expansion) entre le dépôt et le substrat. Pendant la
déposition de revêtement, les contraintes intrinsèques
décroissent avec la température du substrat (figure II.2).
3- Contraintes externes : elles sont dues
aux déformations des revêtement/substrat.
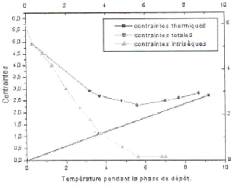
Figure II.2 : Contribution des
contraintes thermiques et intrins èques.
|
Généralité sur les carbures des
métaux de transition.
|
II.3. Préparation des carbures :
à deux seulement : la métallurgie des poudres et la
déposition des couches minces.
II.3.1. Métallurgie des poudres :
Le tableau II.3 résume les méthodes
utilisées en métallurgie des poudres pour préparer les
carbures polycristallins [2,43]. La réaction directe des deux
éléments (métal en poudre et carbone) se fait par fusion
ou frittage. La consolidation des carbures sous des formes utiles se fait par
frittage (puis traitement thermique) ou par compression chaud.
Tableau II.3 : Méthodes de
préparation de carbures.
|
Méthodes
|
Réactions
|
|
a)- Réaction directe des
éléments dans une atmosphère protectrice ou sous
vide.
|
M+C MH+C
|
2
|
b)- réaction
avec le carbone en excès dans une
atmosphère protectrice ou réductrice.
|
MO + C
|
|
c)- réaction du métal avec un gaz
organique
|
M + CxHy C + H2
M + CO 2
|
d)- métal en
présence
|
MCl4 +CxHy + H2
|
mHn)
|
e)- décomposition du métal carbonyle
en
|
M + H2
|
2, H2, H2O)
|
f)- électrolyse des sels
fondus
|
|
Na2O B2O3 + NaCO3 +LiF + MoO3
|
|
II.3.2. Déposition des couches minces:
Des couches minces de carbures peuventêtre obtenues par
dépôt direct des carbures sur
de carbures. Elle
consiste à déposer une couche de métal pur
sur des substrats en acier riche en carbone puis
carbone du substrat vers la couche superficielles du métal
et par la suit sa liaison avec les atomes métalliques [57].
|
Généralité sur les carbures des
métaux de transition.
|
La différence fondamentale entre le
procédé de déposition avec introductions de gaz
réactifs (N2, CH4 de l'adhérence des couches au substrat. En
effet, la faible adhérence du film à son substrat,
l'inhomogénéité de la composition et la présence de
gaz explosif (toxique) sont les principaux problèmes posés lors
de la déposition directe de carbure ou de nitrure de métaux de
transition par PVD ou CVD. Alors que dans le second procédé,
l'interface n'existe pratiquement pas, vu l'interdiffusion des
éléments du substrat dans le film. On obtient ainsi, une
quasi-continuité de la matière du substrat au revêtement et
une adhérence très forte des couches au substrat.
II.4. Carbures de tungstène :
propriétés et applications
Le tungstène pur est un métal de transition de
couleur allant du gris acier au blancétain. Il a le plus haut point de
fusion (3422 °C) et la plus grande résistance de traction à
une température supérieure à 1650 °C. Sa
résistance à la corrosion est excellente, et il forme une couche
protectrice en oxyde lorsqu'il est exposeà l'air [58]. Le
tungstène peut être employé comme un métal pur ou
mélangé à d'autres métaux pour faire des alliages
[59].
Le carbure de tungstène est l'un de ces alliages, il
est composé d'un alliage de tungstène et de carbone qui est
obtenu par une suite de réaction chimique. Puisque son point de fusion
est d'environ 2800 °C, il est impossible de mettre en forme par fusion
[60]. La formation
plusieurs phases avec la dépendance de la structure et la
composition de ces phases avec les paramètres du procédé
suivi et le type de substrat utilisé [46].
Après une centaine d'année de la
découverte du tungstène en 1783. Henri Moissan (1852-1907) a pu
former le carbure de tungstène W2C en 1896, au laboratoire de
l'école de pharmacie à l'université de paris, et en 1898,
P. Williams a formé le carbure WC, alors que le premier carbure de
tungstène fritté a été produit en 1914 [61].
II.4.1. Propriétés structurale
:
Les métaux du groupe VIA forment des carbures de forme
M2C et MC. Le système W-C a été étudié par
Rudy [62]. Il comporte deux composés : le WC se cristallise en structure
hexagonal simple, et son paramètre de maille est a = 2.9065 Å, c =
2.8366 Å; tandis que le W2C a une structure hexagonal compacte [63].
Le diagramme de phase du système W-C en fonction de la
température et du pourcentage de carbone est représenté
sur la figure II.3. En consultant ce diagramme, on
|
Généralité sur les carbures des
métaux de transition.
|
constate que le système W-C présente trois grandes
zones. La première contient la phase W2C, la deuxième contient la
phase WC1-X, et la troisième contient la phase WC.
La première phase W2C se cristallise à une
structure hexagonale compacte (L' 3) (figure II.4-a) où les
atomes de carbone occupent la moitëdes sites interstitiels
octaédriques disponibles [63]. Cette phase apparaît sous trois
formes allotropique [62]: hexagonal commandé entre 1500 et 2450 K,
orthorhombique entre 2450 et 2750 K, et Hcp entre 2750 et 3050 K. La
deuxième phase WC1-X se cristallise en un réseau
cubique à face centrée (B1) (figure II.4-b) où les atomes
de carbone occupent tous les sites interstitiels octaédriques du
réseau du métal, et donnent lieu à un deuxième
réseau CFC inséré dans le premier. Tandis que, la
troisième phase WC se cristallise à une structure hexagonale
simple (figure II.4-c) où les atomes de carbone prennent les positions
(1/3,2/3,1/2) dans la maille unitaire.

100 90 80 70 60 50 40 30 20 10 0

C at.%.C W
Figure II.3 : Le diagramme de
phase du système W-C en fonction de la température et
du
pourcentage de carbone.
|
Généralité sur les carbures des
métaux de transition.
|
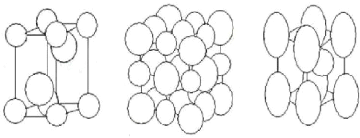
a) W2C b) WC1-x c)
WC
Figure II.4 : Les
différentes structures cristallines des carbures de tungst ène
[64-65].
La phase W2
34%.at C à 2715 °C. C eutectoide entre W et
WCà 1250 °C
et fond avec la solution solide de W à (2715
#177; 5) °C et avec WC1-x approximativement à
2758 °C [66]. L 2C sont obtenues comme des
produits
intermédiaires pendant la production de WC. La phase WC est
la seule phase binaire stableà
la tempé °C [66]. La figure II.5
montre les
positions atomiques de C et W dans les deux phases WC et W2C [67].
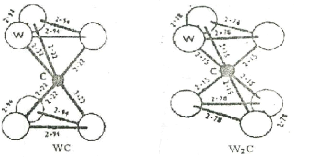
Figure II.5: Les positions atomique
du carbone et de tungst ène dans les phases WC et W2C
[67].
|
Généralité sur les carbures des
métaux de transition.
|
II.4.2. Propriétés physiques et
mécaniques:
Les carbures de tungstène sont des matériaux
durs et très stables à haute température, ils sont
caractérisés par: une dureté relativement grande
atteignant (2200 Kg/mm2 et 3000 Kg/mm2) est
observée pour les deux carbures WC et W2C respectivement (ces
composés sont extra durs par rapport au métal de base W (360
Kg/mm2)), une température de fusion très
élevée ( une résistivité plus élevée
(22 u ène
(5.39 u Elle lui permet classé parmi les
matériaux conducteurs [42], un bas coefficient de dilatation thermique
(5.2mm/k), un module d'élasticité extrêmement important et
une bonne conductivité thermique. Ces deux dernières
propriétés sont avantageuses dans les outils de coupe. Ces
revêtements sont également fortement résistants à la
corrosion dans des milieux acides. Le tableau II.4 représente les
propriétés physiques et mécaniques de tungstène et
de carbure de tungstène.
Tableau II.4 :
Propriétés physiques et mécaniques de
tungst ène et de carbure de tungst ène.
|
Propriétés
|
Tungstène
|
Carbure de tungstène
|
|
Formule moléculaire
|
W
|
WC
|
|
Structure cristalline
|
Cubique Centré
|
Hexagonale simple
|
|
Paramètre de maille :
a( Å ) ; c ( Å
)
|
a =3,165
|
a =2.9065
c =2.8366
|
|
Dureté (kg/mm2)
|
360
|
2200
|
|
Point de fusion
(°C)
|
3680
|
2870
|
|
Module de Young (GPa)
|
407
|
720
|
|
Conductivité thermique
(W·m 1·K
1)
|
174
|
84.02
|
|
Résistivité (u .cm)
|
5,39
|
22
|
|
coefficient de dilatation
thermique
(10-6K-1)
|
4,59
|
5.2
|
|
Densité (g/cm3)
|
19,3
|
15.8
|
|
Couleur
|
gris acier au blanc étain
|
gris-noir
|
|
Résistance à la traction
(GPa)
|
/
|
0.3448
|
|
Généralité sur les carbures des
métaux de transition.
|
II.4.3. Application de carbure de
tungstène
Grâce à leurs propriétés remarquables,
Les revêtements durs en carbure de tungstène sont très
importants dans l'industrie et les diverses applications technologiques. Jls
sont
de composants à haute température dans les
domaines ëronautique et nucléaire [37-39]. Les carbures de
tungstène utilisés aussi comme des couches ésistants
à la corrosion, à l'érosion et à rayer pour des
bijoux. Ces derniers sont également utilisés dans la fabrication
des abrasifs et des roulements. En plus, les carbures de tungstène sont
également trouvés comme les métaux nobles tels que Pt, Pd
et Jr dans l'industrie de catalyse (comme catalyseurs dans les éactions
de synthèse d'hydrogénation, de méthanisation et
d'ammonium). En raison de leurs basses ésistivités
électriques et leurs stabilités chimiques et thermiques, les
couches minces des carbures de tungstène sont utilisées comme
barrière d microélectronique [68].
|
Procédures expérimentales et méthodes de
caractérisation.
|
Dans ce chapitre, nous décrirons les procédures
expérimentales suivies durant notre travail, puis nous décrirons
les techniques de caractérisation.
III.1. Préparation des échantillons
:
que étude expérimentale, en raison de son
influence directe sur les propriétés recherchées. Elle se
déroule en plusieursétapes :


La préparation des substrats
:
surface des substrats utilisés, ainsi que les
différents types de nettoyage qui précèdent
s planes et propres.
Le dépôt des couches minces suivant
une m éthode appropriée.
III.1.1.Préparation des substrats :
Les aciers à coup rapide sont
caractérisés par une dureté élevée à
la température ambiante, comprise entre 60 et 70 HRC et par une
résistance à l'adoucissement élevée. Leurs teneurs
en carbone sont supérieures ou égales à 0.7 % en masse.
Dans le cadre de notre étude on a utilisé des substrats d
Le par une tronçonneuse (avec un disque en SiC
très
fin) sous forme de petites épaisseur de 2mm,
cette découpe se fait sans échauffement des bords
des substrats.
Tableau III.1 : La composition
chimique des aciers XC70.
|
Norme Afnor
|
C %
|
Si %
|
Mn %
|
S %
|
P %
|
|
XC70
|
0,65 - 0,73
|
0,15 -0,35
|
0,40 0,70
|
0,035
|
0,035
|
Le tableau III.1 représente la composition chimique de
ces substrats. La nature des substrats, ainsi que leur état de surface,
influent énormément sur les propriétés physiques de
dépôt. Pour cela ces substrats ont subi (avant la
déposition des couches minces de tungstène) les traitements
mécaniques et chimiques suivants:

Polissage mécanique.
Nettoyage chimique et ionique.
|
Procédures expérimentales et méthodes de
caractérisation.
|
|
a)- Polissage mécanique :
conditions métallographiques standard (le
dégrossissage et le finissage) la première
étape
consiste à polir grossièrement la surface sur des
papiers sablés (en SiC) en rotation à pouvoirs
a seconde étape consiste à finir le polissage en
appliquant la pièce sur des papiers de :


Le dégrossissage : il
se fait avec des papiers abrasifs de grains de plus en plus fins. On a
utilisé les papiers de numéro : (100, 240, 500, 800, 1000),
montés successivement sur un touret horizontal tournant à une
vitesse constante. Le polissage se fait , tout en le remuant dans le sens
opposé à la rotation du plateau. Ceci est fait sous
coulée
Ce type
adhèrent fortement et résistent en particulier
à une attaque chimique [68]. Il permet

éviter tout au long
papier différent.
Le finissage : On termine le
polissage des substrats par un polissage de précision en les
: la première est la plus grossière
et la dernière est la plus fine) soit la patte
diamantée.
b)- Nettoyage chimique et ionique :
Après le polissage mécanique, les substrats
subissent un nettoyage (dégraissage) pour éliminer les
contaminations organiques (graisse, poussières, etc.).
distillée
suivants :

Dégraissage pendant 10 min dans un bain de
trichloréthylène activé aux ultrasons. Lavage pendant 10
min dans
Lavage pour une deuxième fois pendant 10 min dans un bain
de méthanol activé aux ultrasons.
|
Procédures expérimentales et méthodes de
caractérisation.
|
Enfin, et juste avant la déposition de la couche de
tungstène, on expose les substrats à un
ple [69] pendant 10
min. Le bombardement est in-situ sous
une pression de 1Pa et avec une
tension de 1000V et un courant de 100mA, pouréliminer tout

organiques) et donne des surfaces très propres et
très lisses.
On évite de toucher la surface du substrat,
pouréviter toute contamination.
III.1.2. Préparation du dépôt de
tungstène:
/
XC70). Les couches minces de tungstène sont
déposées par la technique de pulvérisation cathodique (RF)
dans un vide de 10-7mbar. Les conditions du dépôt des
couches minces de tungstène pour les trois séries, sont
résumées ci-dessous (tableau III.2) : Les températures
de
quelques centimètres. La puissance de décharge a
été maintenue constante à 2000 W, la
Å/min. La distance entre la cible et les
substratsétait fixée à 150 mm, on peut noter que le temps
total de dépôt dans les trois séries est différent
d'une série à l'autre: c'est-à-dire on a
élaboré des échantillons de différentes
épaisseurs pour les couches minces de tungstène, la
première série d'échantillons (d'épaisseur 2um), la
deuxième série (d'épaisseur 4um) et enfin la
troisième série (d'épaisseur 6um).
Tableau III.2: Les conditions
expérimentales de dépôt des couches minces de tungst
ène.
|
Série 01
|
Série 02
|
Série 03
|
|
La cible
|
Tungstène
|
Tungstène
|
Tungstène
|
|
Vide (mbar)
|
10-7
|
10-7
|
10-7
|
|
température de dépôt (°C)
|
500
|
500
|
500
|
|
La puissance de décharge (W)
|
2000
|
2000
|
2000
|
|
La vitesse de déposition (A°/min)
|
1350
|
1350
|
1000
|
|
Distance entre la cible et le
substrat (mm)
|
150
|
150
|
150
|
|
temps de dépôt (min)
|
15
|
30
|
60
|
|
Epaisseur de la couche (pm)
|
|
|
|
|
Procédures expérimentales et méthodes de
caractérisation.
|
III.1.3. Les traitements thermiques appliqués
(recuits) :
Afin de provoquer la réaction entre les constituants
des échantillons (W, Fe, C), et par conséquent la formation des
carbures à l'interface substrat/revêtement, les
échantillons (couches minces/substrats) ont subis des
traitements thermiques (recuits) sous vide secondaire pour éviter
l'oxydation. Ces traitements sont effectués à diverses
températures et pendant différentes durées comme
indiqué dans le tableau III.3 suivant:
Tableau III.3 : Différents
recuits thermiques utilis és.
|
Température de recuit Tr (°C)
|
600
|
700
|
800
|
800
|
900
|
1000
|
|
Temps de recuit tr (min)
|
30
|
30
|
30
|
60
|
30
|
30
|
|
Vide utilisé (torr)
|
10-7
|
|
Température de recuit Tr (°C)
|
800
|
800
|
900
|
1000
|
|
30
|
60
|
30
|
30
|
|
Temps de recuit tr (min)
|
|
10-7
|
|
Vide utilisé (torr)
|
|
Température de recuit Tr(°C)
|
600
|
700
|
800
|
900
|
1000
|
|
Temps de recuit tr (min)
|
30
|
30
|
30
|
30
|
30
|
|
10-7
|
|
Vide utilisé (torr)
|
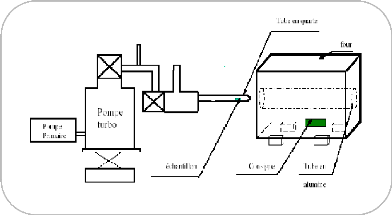
Figure III.1 : Montage
expérimental du système de recuit utilisé.

2 d hkl .sin( hkl ) n
. (III.1)
Avec : hkl l'angle entre le faisceau incident et les
plans diffractant d'indices de Miller (hkl).
dhkl la distance interréticulaire entre les plans
diffractant.
la longueur d'onde du faisceau incident.
n de la diffraction.
, les taux de défauts structuraux, les macro et micro-
0].
|
Procédures expérimentales et méthodes de
caractérisation.
|
Les traitements thermiques de nos échantillons ont
été faits au laboratoire de physique
niversité Farhat
Abbas - Sétif. Les échantillons sont introduits dans un tube en
quartz
(figure III.1) raccordé à un groupe de pompage,
constitué de deux pompes primaire et
secondaire, destiné
à évacuer l'air jusqu'à une pression égale
10-7 torr. Ce tube est chauffé à
de type F21130
(modèle 21100). La figure III.1 montre le montage
expérimental du système de recuit
utilisé.
III.2. Les méthodes de caractérisation
:
Le but de ce travail est l'étude des
propriétés structurales et mécaniques des
revêtements en carbures de tungstène formés à partir
d'une couche mince de tungstène déposée sur des substrats
en acier XC70, par pulvérisation cathodique radiofréquence
à effet magnétron. Donc

des plusieurs facteurs de dépôt qui sont entre
autres: le substrat, , les paramètres de dépôt, la
polarisation du substrat, la concentration du carbone dans le substrat, la
concentration et la vitesse du gaz réactif. ces buts que ces couches ont
été analysées par différentes techniques de
caractérisation des matériaux.

variées:

La diffraction des rayons X (DRX) pour déterminer la
structure, la taille de grain et les contraintes.
Le microscope optique et la microscopie électronique
à balayage pour observer la morphologie de la surface.
Les essais Vickers pour mesurer la micro-dureté de ces
revêtements.
Nous présentons les différentes techniques
auxquelles nous avons eu recours pour caractériser notre
matériau.
III.2.1. Diffraction des rayons X (DRX).
La diffraction de rayons X aété utilisée
pour caractériser la structure cristallographique des
échantillons et mettre en évidence les différentes phases
cristallines présentes dans les
phases au cours de divers traitements notamment des recuits.
Par ailleurs, la diffraction des rayons X sur la matière cristalline
permet d'avoir accès à des informations physiques sur les
cristaux, notamment leur taille et leur orientation.
|
Procédures expérimentales et méthodes de
caractérisation.
|
III.2.1.1. Le principe :
polycristallin, il est en partie réfléchi par les
plans atomiques de certains cristaux. Pour que la
atomiques doit avoir lieu sous un angle particulier. Il faut
en effet que les ondes réfléchies soient en phase de sorte
à interférer de manière constructive pour ensuite
être mesurées par le détecteur, comme il est
illustré à la figure. III.2.
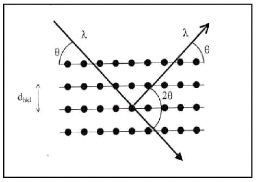
Figure III.2 : Famille de plans
cristallins en condition de Bragg.
Les conditions nécessaires
à cette interférence constructive sont données par la loi
de Bragg:
|
Procédures expérimentales et méthodes de
caractérisation.
|
Les positions angulaires des raies de diffraction sont
caractéristiques des paramètres du
permet donc de remonter au
déterminé, les positions angulaires des raies
permettent de calculer les distances interréticulaires des plans
atomiques diffractant et a
Les positions et intensités des raies de diffraction de
la plupart des matériaux connus ont
diffractogramme expérimental avec ces données
permet de retrouver la nature de chaque
III.3.1.2. Les conditions expérimentales :
L'enregistrement des spectres, d'intensité I
diffractées en fonction de par rapport à la surface de
l'échantillon, est effectué par un diffractomètre de
poudre, automatisé Siemens de type (BRUKER-AXS type D8) (figure III.3)
opérant en géométrie Bragg - Brentano, suivant les
conditions suivantes:

Figure III.3 :
Diffractomètre de type (BRUKER - AXS type D8).

|
Procédures expérimentales et méthodes de
caractérisation.
|

La source des rayons X est produite par une anticathode cuivre,
alimentée par un générateur tension - courant de 35 kV 30
mA.

Le rayonnement X utilisé est K du cuivre ( Cu =
1.54056Å) obtenu par un monochromateur germanium.
L'échantillon est placé sur une tête
goniométrique.
Les spectres de DRX des échantillons sont
enregistrés pour 2 compris entre 20° et 120° avec un
pas de 0.02°.
L'identification des phases présentes dans les spectres de
DRX a été faite en utilisant les fichiers ASTM.
III.3.1. 3. Détermination de la taile des
gains:
Dans un premier temps, il est possible,à partir des
spectres de diffraction, de calculer la taille des cristallites des phases des
dépôts en utilisant la formule de Scherrer [71-72] donnée
par la relation III.2. Celle-ci ne tient pas compte des effets des
microdéformations sur la
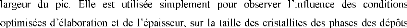
considérés.
0,9 .
D
.cos
Où :
à mi-
(figure III.4) ; en anglais FWHM (Full width half
maximum).
D est la taille moyenne des cristallites ([D] = nm).

est l'angle de diffraction en degrés.
est la longueur d'onde du faisceau de rayon X.
|
Procédures expérimentales et méthodes de
caractérisation.
|
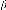
Figure III.4: Illustration
montrant la définition de à partir de la courbe
de diffraction
des rayons X.
III.3.2. La microscopie électronique à
balayage (MEB).
Le microscope électronique à balayage (MEB)
(scanning electron microscopy-SEM en anglais) a été
imaginé pour la première fois en Allemagne, dans les
années 1930, par Knoll et Von Ardenne et développé par
Zworykin, Hillier et Snyder dans les laboratoires RCA aux Etats-Unis (1940).
Mais La microscopie électronique à balayage a connu son
véritable essor entre 1948 et 1965, grâce aux progrès
techniques de la télévision et des détecteurs
d'électrons et grâce aux recherches d'Oatley et de ses
condisciples à Cambridge. Cette nouvelle technologie a permis, du fait
de sa profondeur de champ, l'observation du relief d'échantillons
massifs.
III.3.2.1. Le principe :
Les microscopes à balayage utilisent un faisceau
d'électrons très fin qui balaye point par point la surface de
l'échantillon. L"interaction du faisceau avec l'objet crée
différentes émissions de particules qui sont analysées
à l'aide de détecteur approprié : électrons
secondaires, électrons rétrodiffusés, électrons
transmis, émission de rayons X (figure III.5). Captée de
façon synchrone avec le balayage du faisceau sur l'échantillon,
l'intensité de chacun de ces signaux module la luminosité
d'écran de télévision donnant des images noir et blanc que
l'on peut mixer entre elles.
|
Procédures expérimentales et méthodes de
caractérisation.
|
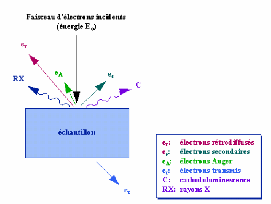
Figure III.5 :
Représentation schématique de l'interaction
entre un faisceau d' électrons et la
surface d'un
échantiion.
Le principe de fonctionnement d'un
microscopeélectronique à balayage ayant un filament de
tungstène comme source d'extraction desélectrons est
décrit dans le schéma cidessous (figure III.6).
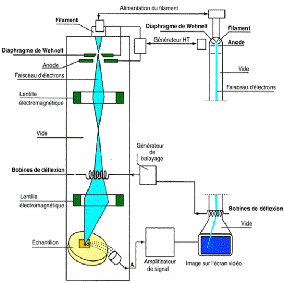
Figure III.6 :
Représentation schématique du principe de
fonctionnement du MEB.

|
Procédures expérimentales et méthodes de
caractérisation.
|
III.3.2.2. Les conditions
expérimentales:
Dans ce travail, pour étudier la topographie de la
surface de noséchantillons, nous avons utilisé un microscope
électronique à balayage (MEB) de la marque Philips XL 30,
opérant avec une t KV.
III.3.3. Le microscope optique :

détails in
III.3.3.1. Le principe :
Le microscope optique se base sur les lentilles pour obtenir une
image agrandie de

On peut faire un microscope simplifié (figure III.7)
avec deux lentilles convergentes. objectif ». - delà de la distance
focale, cela forme une image réelle inversée et de taille
différente
, si celui-ci est situé à une distance
inférieure au double de la
: elle est positi

», donc en relâchant les muscles
Figure III.7: Le principe du
microscope optique.
|
Procédures expérimentales et méthodes de
caractérisation.
|
III.3.3.2. Les conditions expérimentales
:
Le microscope optique est un appareil qui donne la
micrographie des surfaces des échantillons. Il donne des informations
telles que la répartition de nouvelles phases, la taille des grains, les
joints de grains, les fissures, etc. L'observation de noséchantillons
(W/acier) recuit et non recuit est effectuée par un microscope optique
de type EURO-MAX microscope (Hollande) relié à un
micro-ordinateur pour faciliter le traitement des images. Les grossissements
varient entre 10 et 400.
III-3-4. La micro-dureté:

Figure III.8 :
; on mesure les deux diagonales d1 et d2
On obtient la valeur d en effectuant la moyenne de d1 et d2
es.
50
La dureté d'un métal est sa capacité
à résister à la déformation. On la détermine
au moyen d'essais normalisés (Brinell, Meyer, Vickers, Rockwell).
Le principe est toujours identique : un pénétrateur
indéformable laisse une empreinte dans le matériau à
tester (figure III.8). On mesure les dimensions de l'empreinte et on en
déduit la dureté. Dans notre étude les mesures de la
micro-dureté ont été réalisées avec un
indenteur de type Vickers.
|
Procédures expérimentales et méthodes de
caractérisation.
|
F N
( )
HV 0,1 02 .... (III.3)
S mm
( 2)
Où F est la force appliquée par la charge P, donc
elle estégale à :
F 9,8 1 P(Kg) .

et S est l . Elle
d du carré de la

2
d
S
2.
sin 68
0, 1891

HV
F

d
2
HV
P
1,8 54
d
2
Alors :
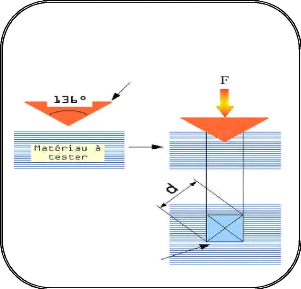
Pénétrateur : pyramide en diamant de base
carrée et d'angle au sommet entre 2 faces opposées égal
à 136°
Empreinte
Figure III.9: L'indenteur et
l'empreinte d'indentation.
|
Procédures expérimentales et méthodes de
caractérisation.
|
III.3.4.2. Profondeur de pénétration de
l'indenteur dans un essai d'indentation :
La profondeur de pénétration h est facilement
calculée ; h peut être exprimée en fonction de la diagonale
d de la base carrée de l'empreinte pyramidale (dans le cas d'essai
Vickers). La profondeur h estégale à :
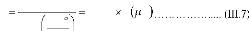
d
h 0.202
2 tan
136
dm

2
Quand une charge importante est appliquée, l'indenteur
pénètre profondément dans l'échantillon. Si la
profondeur de pénétration dépasse l'épaisseur du
revêtement, le pénétrateur continue son enfoncement dans le
substrat. La forme de la zone déformée ne correspond plus
à la forme de l'indenteur, si la dureté du substrat
diffère considérablement de celle du revêtement
III.3.4.2. Les conditions expérimentales
:
Les essais de la micro-
microduromètre de type FM-300e de la socëté
Futur_TECH_CORP (Japon) avec un pénétrateur en diamant sous forme
pyramidaleà base carrée. Les charges utilisées sont : 10g,
25g, 50g, 100g, 200g, 300g, 500g, et 1000g.
|
Résultats et interprétations.
|
Dans la première partie de ce chapitre nous
présentons les résultats de notre travail portant sur la
caractérisation des échantillons recuits et non recuits.
Leséchantillons constituant la série couche mince de
tungstène 6um déposée sur un


XC70 et notés dans la suite (W/XC70). En plus, la
deuxième partie présente une étude comparative entre les
trois séries (S1, S2 et S3). de sur les
différentes
propriétés structurale, morphologique et
mécanique a été étudiée.
IV.1. Caractérisation des couches minces de la
série S3 :
IV.1.1. Etudes structurale et morphologique :
La structure cristalline des couches minces dépend de
plusieurs facteurs notamment le vide,
paramètres jouent un rôle très important dans
la détermination de la structure des couches minces.
Dans ce contexte, on a obtenu quelques diagrammes de diffraction
des rayons X réalisés sur le substrat XC70 nu et sur
deséchantillons (W/XC70) de la troisième série avant et
après recuit.
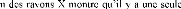
IV.1.1.1. Analyse des échantillons non recuits et
recuits entre 600 et 700°C : L'analyse des substrats XC70 nus par
diffractio
phase Fe qui diffracte, avec une absence totale du carbone et de
ses alliages (figure IV.1). Les résultats de l'analyse des substrats
revêtus par une couche mince de tungstène, d'épaisseur 6
avant tout recuit thermique sont illustrés dans la figure IV.2. On
observe l'existence de cinq pics seulement situés à : 40.36°
; 58.36°; 73.16°; 100,64° et 114.88°. Ces pics sont
attribués à la phase W et correspondent aux familles de plans
(110), (200), (211), (310) et (222) respectivement. On remarque
également l'absence des pics du substrat. Ceci est dueévidemment
à l'épaisseur relativement grande de la couche mince de
tungstène par rapport à la profondeur maximale de
(Cu)=1.5406Å est u=0.33um-1 [58]).Il est
intéressant, par ailleurs, de noter la croissance
privilégiée suivant la direction [222] au lieu de la direction
[110] d'après les fichiers ASTM du tungstène. En outre, il faut
souligner l'apparition des quelques pics en plus de ceux du tungstène
dans les spectres de DRX enregistrés pour les échantillons
recuits à 600°C et 700°C
|
Résultats et interprétations.
|
(Figure IV.3). Ces pics correspondentà la phase Fe
(ferrite) et la phase Fe3C (Cémentite). L'observation de ces
réflexions est due probablement au décollement partiel de la
couche mince de tungstène. La non détection de carbures de
tungstène à ces températures nous laisse penser que
l'apport énergétique n'est pas suffisant pour activer la
diffusion des atomes du carboneà travers le substrat vers la couche de
tungstène, afin de former ces carbures.
de diffraction correspondant aux différentes phases
constituant les échantillons, avant et après
le fichier ASTM (Annexe).
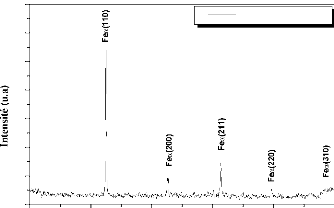
70
Substrat XC70
60
50
40
30
20
10
0
20 40 60 80 100 120
2 (degrés)
Figure IV.1 : Spectre de DRX du
substrat XC70.
|
Résultats et interprétations.
|
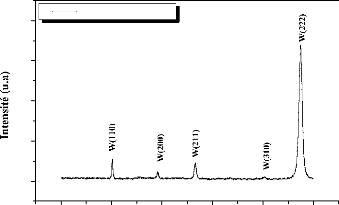
200
150
100
50
0
Ech S3 non recuit
20 40 60 80 100 120

2 (degrés)
Figure IV.2 : Spectre de DRX de 6
m)/XC70] non recuit.
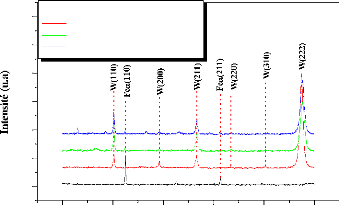
0
300
250
200
150
100
50
substrat XC70
Ech S3 non recuit
Ech S3 recuit à 600°C/30min
Ech S3
recuit à 700°C/30min
20 40 60 80 100 120

2 (degrés)
Figure IV.3 : Spectres de DRX des
échantillons [W (6 m)/XC70] recuits
à 600 °C et 700
°C pendant 30 min.
|
Résultats et interprétations.
|
PV.1.1.2. Analyse des échantillons recuits entre
900 et 1000°C :
Le recuit des échantillons entre 900°C et
1000°C pendant 30 minutes (figure IV.4) favorise l'interdiffusion et
provoque la réaction entre la couche mince de tungstène et le
substrat, ce qui conduità la formation de nouvelles phases.
20 40 60 80 100 120
2 (degrés)

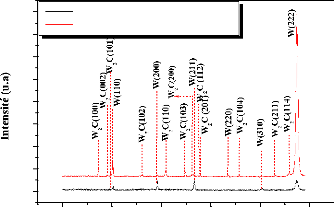
400
350
300
100
50
0
Ech S3 Tr=900°C ; tr=30min) Ech S3
Tr=1000°C ; tr=30min)
250
200
150
Figure IV.4 : Spectres de DRX des
échantillons de la série S3,
recuits à 900 et 1000°C pendant 30 min.
Le spectre de DRX enregistré pour un échantillon
recuit à 900°C pendant 30 minutes, montre pic, en plus de ceux du
tungsêne. Il est identifié comme étant du carbure W2C. Ce
pic situé vers ~40°, correspond à la famille de plan (101)
de la phase hexagonale W2C. On remarque aussi à cette
température, une chute drastique de l'intensiédu pic de
tungstène pur situé à 114,88°.
Par contre, pour un recuit à 1000°C pendant 30
minutes, il apparait une série de 12
nouveaux pics situés
à 34,5°, 38,09°, 39,61°, 52,24°, 61,88°,
69,61°, 72,4°, 74,79°, 75,76°,
91,45°, 106,27°
et 112,02°, en plus de ceux du tungsêne. Ces nouveaux
pics,étant aussi attribués
à la phase W2C,
correspondent aux familles de plans (100), (002), (101), (102), (110),
(103),
(200), (112), (201), (104), (211) et (114) respectivement. La
formation de ce carbure résulte
re le carbone du substrat et le
tungstène du revêtement (le carbone étant
|
Résultats et interprétations.
|
IV.1.1.3. Evolution de la structure de la couche de
tungstène avec la température de recuit :
Dans l'ensemble des spectres de diffraction des rayons X relatifs
auxéchantillons avant et après recuit, on remarque que la couche
de tungstène est polycristalline et présente une
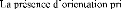
orientation préférentielle suivant la direction
[222]. vilégiée dans les
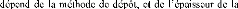
films minces couche [73-75]. Le recuit
thermique provoque la formation et la croissance du carbure W2C
de tungstène, il en résulte la
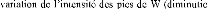
n) et de W2C (augmentation). Cependant, il est
intéressant de souligner l'augmentation remarquable de
l'intensité du pic (222) de tungstène pur situé à
114,88° (figure IV.4). Ceci est du, probablement,
de la structure de la couche de tungstène restante.
En effet, conformément au tableau JCPDS-ICDD (Annexe),
la famille de plans (222) du tungstène massif diffracteà
=114,917°. En ce qui concerne notre cas, et pourl'échantillon non
recuit, ce pic se trouve à 114.88°. En revanche, dans les autres
échantillons recuits à des températures 600°C et
700°C, on note un déplacement de ce pic vers les angles 2
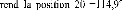
=115.24° respectivement. Mais ce pic p à 900°C,
comme il est
montré dans la figure IV.5.
Ech S3 non recuit
Ech S3 Tr=600°C , tr=30min Ech S3 Tr=700°C ,
tr=30min Ech S3 Tr=900°C , tr=30min Ech S3 Tr=1000°C ,
tr=30min
500
400
300
200
100
0
800
700
600
110 112 114 116 118 120

2 (degrés)
Figure IV.5 : Evolution du pic
(222) de tungstène avec la température de recuit.
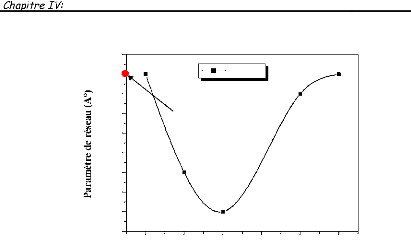
Résultats et interprétations.
Ech S3
3,165
3,164
Paramètre de réseau de
Wmassif
3,163
3,162
3,161
3,160
3,159
3,158
500 600 700 800 900 1000
3,167
3,166
Température de recuit (°C)
Figure IV.6 : Evolution du
paramètre de réseau avec la température de recuit.
|
|
Taille de grain
|
|
|
|
15 14 13 12 11 10 9 8 7
|
500 600 700 800 900 1000
Tr (°C)
Figure IV.7 : Evolution de la
taille de grains avec la température de recuit.
|
Résultats et interprétations.
|
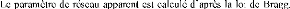
à partir de la position
angulaire du pic (222). La variation de ce paramètre en
fonction de la température de recuit est
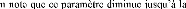
illustrée dans la figure IV.6. O température de
recuit
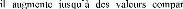
Tr=700°C, puis able à celui de tungstène
massif
(a=3.165A°) pour les températures
supérieures.
On a ensuite calculé la taille de grains à
partir de la largeur à mi-hauteur du pic (222) en utilisant la formule
de Scherrer (voir chapitre III). L'évolution de la taille des grains
avec la température de recuit est représentée sur la
figure IV.7. Comme il est prévisible, la taille des grains croit entre
500°C et 700°C on

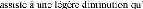
est due probablement au passageà la recristallisation.
La croissance de la taille des grains avec
l'élévation de la température de recuit, permet de dire
que le recuit dans l'intervalle consiáré entre 700 et
1000°C, offre l'énergie nécessaire aux atomes de
tungstène pour se réarranger dans le réseau cristallin et
se placer dans des sites de substitution normaux.
IV.1.2. Etudes microstructurale et morphologique
:
a)-Observation par microscope optique :
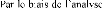
par microscopie optique deséchantillons non recuits, on
constate que ces derniers possèdent des surfaces relativement lisses
avec une couleur marron bâlant (figure. IV. 8-a).
.
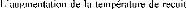
fait évoluer la morphologie de la surface. Par exemple :
pour un recuità 900°C pendant 30 minutes (figure. IV. 9-a)
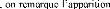
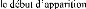
de taches de couleur gris métallique,
aléatoirement parsemées sur toute la surface, ce qui confirme la
formation du carbure de tungstène W2C. On constate, également
à cette température, de fissures à la surface des
échantillons.
|
Résultats et interprétations.
|

a
Gx100
b
Gx100
Figure IV.8 : Micrographies
obtenues par microscopie optique des échantillons
[W (6 m)/XC70]: a)-
non recuit et b)- recuit à 700°C pendant 30min.

a
Gx10
b
Gx10
Figure IV.9: Micrographies obtenues
par microscopie optique des échantillons [W (6 m)/XC70]: a)- recuit
à 900°C pendant 30min et b)- recuit à 1000°C pendant
30min.
|
Résultats et interprétations.
|

Gx400
Figure IV.10 :
Micrographie obtenue par microscop
[W (6 m)/XC70]: recuit à 1000°C pendant
30min.
la figure IV.9-b. On remarque dans ce cas, la croissance des
taches grises pour former des particules sphériques, et une augmentation
remarquable des fissures (figure. IV. 10).
b)-Observation par microscopie électronique
à balayage (MEB) :
Pour suivre les changementséventuels de la morphologie
deséchantillons, avec un peu plus de détails, on a utilisé
la microscopie électronique à balayage. La figure IV.11, illustre
des images de la surface des échantillons de la série S3 obtenues
par MEB. Comme pour les observations au microscope optique, la morphologie de
la surface deséchantillons change avec la température de
recuit.
recuit (figure IV.11-a), la surface des échantillons
est rela de petites particules blanches dispersées arbitrairement.
examen des échantillons recuits à 700°C pendant 30 minutes
montre que la morphologie de la surface deséchantillons devient plus
rugueuse (figure. IV.11-b). Dans le cas de recuit à 900°C pendant
30 minutes (figure IV.11-c), la rugosité de la surface diminue, un petit
fissures à grande distance réparties aléatoirement dans
les échantillons.
On note également un semblant de début de
décollement par endroits.
|
Résultats et interprétations.
|
Les traitements thermiques deséchantillons à
1000°C pendant 30 minutes provoquent une augmentation remarquable des
fissures (figure IV.11-d). Ceci est peutêtre lié à la
réaction entre le substrat et la couche (c.-à-d la diffusion de
carbone vers la couche).
la couche de revêtement. Semble reposer sur le substrat
sans y adhérer.
Par conséquent, on peut dire que, si la température
augmente, on aura une dégradation par endroits du revêtement de
noséchantillons.

c
a b
d
Figure IV.11 :
Evolution de la morphologie de la surface des échantillons [W
(6 m)/XC70] en fonction de la température : a)- non recuit, b)- recuit
à 700°C / 30 min.
c)- recuit à 900 °C/ 30
mind)- recuit à 1000°C / 30 min.
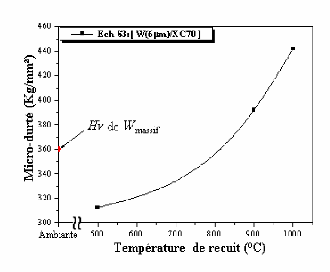
|
Résultats et interprétations.
|
IV.1.3. Mesures de la micro-dureté:
Des essais de micro-dureté ont été
effectués avec des charges variant entre 10g et 1000g. Les variations de
la micro-dureté on été suivies en fonction dela
température de recuit, la charge
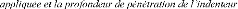
a)-Variation de la micro-dureté en fonction de la
température de recuit :
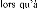
La figure IV.12, présente la variation de la
micro-dureté des échantillons en fonction de la
température de recuit, en utilisant une charge de 50g. Il apparaît
clairement, que la micro-dureté augmente légèrement avec
la température de recuit. La valeur de la micro- chantillon non recuit
est égale 312 kg/mm2 (valeur comparable à celle du
tungstène massif). Cependant, après recuit, les valeurs de la
micro-dureté sont distinctes : par exemple le recuit
deséchantillons à 900°C pendant 30 minutes fait augmenter la
micro-dureté ~400kg/mm2. A 1000°C/30 min, la
micro-dureté devient maximale et prend la valeur ~442kg/mm2.
On remarque aussi que les valeurs de la micro-dureté à toutes les
températures de recuit sont inférieures à celles des
carbures massifs WC (~2200 kg/mm2) et W2C (~3000 kg/mm2)
[45].
Figure IV.12:
Variation de la micro-dureté en fonction de la température de
recuit
|
Résultats et interprétations.
|
La croissance de la micro-dureté des revêtements,
surtout au-delà de 800°C est due, certainement, à la
formation et croissance du carbure W2C. Néanmoins les valeurs obtenues
et qui sont inférieures à celles des carbures massifs trouvant
leur explication dans le réseau des fissures et dans le
quasidécollement du revêtementà ces températures.
b)-Variation de la micro-dureté en fonction de la
charge appliquée :
La figure IV.13, montre les variations de la micro-dureté
des échantillons en fonction des charges appliquées. On remarque
que les courbes ont pratiquement la même allure.les décroissent
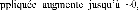
rapidement, lorsque la chargea 3Kg, laquelle on pourrait la

à atteindre la charge de 1Kg
avec des valeurs notamment très proches. La
micro-dureté prend des valeurs, à la surface
des
échantillons, supérieures à celles de volume, et
ceci quelle que soit la température de recuit: A
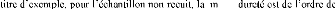
icro- 312Kg/mm2 à la surface; cependant, sur
le palier, c'está-dire en volume, elle est inférieure à
200 Kg/mm2, valeur comparable à celle du substrat. Pour les
échantillons recuits entre 900°C et 1000°C, la
microdureté est considérable à la surface, tandis que sur
les plateaux, elle di
la valeur 200 Kg/mm2. Ceci est dû
principalementà la présence des carbures de tungstène W2C
à l'interface (revêtement/substrat), mis enévidence par
diffraction des rayons X.
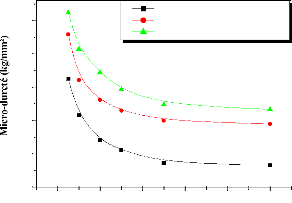
400
Ech.S3.Non recuit Ech.S3.Recuit à 900°C/30min
Ech.S3.Recuit à 1000°C/30min
350
300
250
200
150
0,0 0,2 0,4 0,6 0,8 1,0
Charge (kg)
Figure IV.13:
Variation de la micro-dureté en fonction des charges
appliquées aux échantillons
[W (6 m)/XC70].
|
Résultats et interprétations.
|
c)-Variation de la micro-dureté en fonction de la
profondeur de pénétration :
valeur mesurée ne peut jamais être attribuée
au revêtement seulement. Il faut tenir en compte la
-dureté, on a tracé les
variations de celle-
La figure IV.14, montre les variations de la micro-dureté
en fonction de la profondeur de
-dureté dans tous les cas, diminue en
puis la diminution devient lente. Ce changement en vitesse de
diminution estûdà l'effet du substrat sur la micro-dureté,
et lorsque la profondeur est importante, les valeurs de la microdureté
obtenues se rapprochent beaucoup de celle du substrat. On remarque aussi, que
la profondeur après laquelle la dureté diminue lentement,
dépend de la température de recuit.
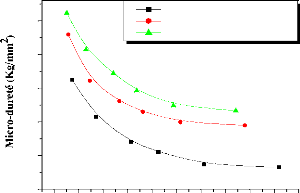
400
Ech.S3.Non recuit Ech.S3.Recuit à 900°C/30min
Ech.S3.Recuit à 1000°C/30min
350
300
250
200
150
2 4 6 8 10 12 14 16 18 20 22
Profondeur de pénétration ( m)

Figure IV.14:
Variation de la micro-dureté en fonction de la profondeur de
pénétration dans
les échantillons [W (6
m)/XC70].
|
Résultats et interprétations.
|
IV.2. Etude comparative entre la série S3 et les
deux autres séries S1 et S2 :
IV.2.1. Etudes structurale et morphologique :
Les spectres de diffraction des rayons X
deséchantillons des trois séries S1, S2 et S3 sont
présentés ci-après. Ils ence du traitement thermique sur
lesévolutions structurale et micro-structurale deséchantillons
étudiés.
Nous avons rapporté, sur la figure IV.15, les spectres
de diffraction des rayons X des échantillons non recuits ble des films,
on constate que les couches minces de tungstène sont polycristallines
avec des orientations préférentielles différentes: suivant
la direction [110] dans la série S1 (d'épaisseur 2um), et suivant
la direction [222] dans les deux autres séries (S2 d'épaisseur
4um et S3 d'épaisseur 6um). On remarque aussi l'absence des pics du
substrat dans les spectres des trois séries. Ceci est dû
certainement aux épaisseurs relativement grandes de ces couches
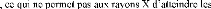
substrats.
L'analyse des spectres de DRX des échantillons recuits
à des températures inférieures à 800°C, ne
montre aucun changement structural par rapportà état non recuit,
sauf qu'on remarque l'apparition de quelques pics du substrat dans les spectres
deséchantillons de la série S3 recuits à 600°C et
700°C. Ceci est dû au décollement partiel de la couche de
revêtement à ces températures. En revanche, le recuit des
échantillons à une température supérieure ou
égale à 800°C (par exemple à 1000 °C/30 min
(Figures IV.16, 17, 18.)), favorise la réaction entre les couches minces
de tungstène et le substrat, et par conséquent la formation des
carbures binaires W2C (pour les trois séries S1, S2 et S3) et WC (pour
la première série), et ternaire Fe3W3C (pour les deux
premières séries).
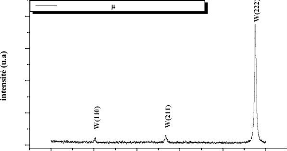
|
Résultats et interprétations.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ech S1 [W (2um)/XC70] non recuit
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ech S2 [ W (4 m )/X C 70 ] non recuit
200
150
100
50
0
20 40 60 80 100 120
2 (degrés)
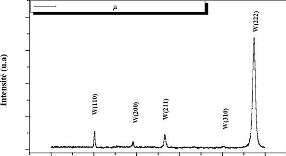
(Ech S3 [ W (6 m )/X C 70 ] non recuit)
200
150
100
50
0
20 40 60 80 100 120

2 (degrés)
Figure IV.15: Spectres de DRX des
échantillons non recuits pour les trois séries.
|
Résultats et interprétations.
|

Figure IV.16: Spectres de DRX des
échantillons de la série S1 [W (2 m)/XC70] avant et
après
recuit [76].
Résultats et interprétations.
Ech S2 non recuit
Ech S2 Tr=800°C , tr=30min Ech S2 Tr=900°C ,
tr=30min Ech S2 Tr=1000°C , tr=30min
350
300
250
200
150
100
50
0
20 40 60 80 100 120
2 (degrés)
450
400
Figure IV.17: Spectres de DRX des
échantillons de la série S2 [W (4 m)/XC70] avant et
après
recuit [77].
Ech S3 non recuit
Ech S3 Tr=900°C ; tr=30min Ech S3 Tr=1000°C ;
tr=30min
500
400
300
200
100
0
20 40 60 80 100 120

2 (degrés)
Figure IV.18: Spectres de DRX des
échantillons de la série S3 [W (6 m)/XC70] avant
et après recuit [présent travail].
|
Résultats et interprétations.
|
IV.2.2. Etudes microstructurale et morphologique
:
La comparaison de la microstructure deséchantillons des
trois séries est donnée par la figure IV.19. Avant recuit, on
remarque que toutes les surfaces deséchantillons sont lisses. Mais
après les traitements thermiques appliqués, la morphologie de
surface change, et devient plus

premières séries S1 et S2. La différence
prin

c
a
b
d
de la série S3 (Fig. IV-d).
Figure IV.19 : La morphologie de
la surface des échantillons avant et après recuit à
900°C
pendant 30 min pour les trois séries a) Non recuit, b)
série 01, c) série 02 et d) série 03.
|
Résultats et interprétations.
|
IV.2.3. Etude de la micro-dureté:
Sur la figure IV.20, nous avons rapporté, pour les trois
séries, la variation de la
micro-dureté en fonction de la
température de recuit. D'après cette figure IV.20, On constate
que
-dureté présente la même allure pour les trois
séries : une augmentation
avec la température de recuit, mais
cette augmentation est légère pour les échantillons de la
série
S3. En plus, les valeurs de la micro-dureté des
échantillons de référence pour les trois séries
S1,
S2 et S3 sont égales respectivement à ~305
Kg/mm2; ~249 Kg/mm2 et ~312 Kg/mm2 (valeurs
comparables à celle du température de recuit, ceci est du
probablementà la formation des carbures.
Par exemple, à 1000°C/30 min la
micro-dureté des échantillons de la séries S1 devient
maximale et atteint ~758 Kg/mm2. Mais pour les autres séries
S2 et S3, la micro-dureté prend les valeurs suivantes ~520
Kg/mm2 et ~442 Kg/mm2.
La variation de la micro-dureté en fonction des charges
appliquées est illustrée sur la figure IV.21. On constate que
toutes les courbes présentent une variation similaire : elles diminuent
lorsque la charge appliquée augmente. Cette diminution prend deux
vitesses différentes , après cette charge, elle devient
très lente avec des valeurs très proches. Les courbes relatives
aux séries S2 et
S3 sont voisines, cependant celle de la série S1 est
nettement décalée vers le haut. Elle présente, par
conséquence, des valeurs de micro-dureté plus grande.
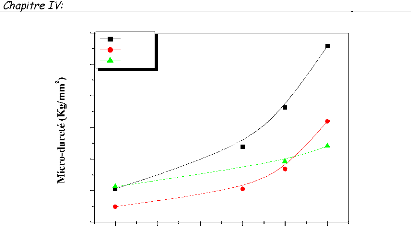
Résultats et interprétations.
Ech.S1.
Ech.S2.
Ech.S3.
400
600
500
300
200
500 600 700 800 900 1000
800
700
Température de recuit (°C)
Figure IV.20: Variation de la
micro-dureté en fonction de la température
de recuit pour les
trois séries.
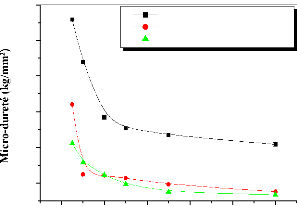
800
700
Ech.S1. recuit à 1000°C/30min
Ech.S2.
recuit à 1000°C/30min
Ech.S3. recuit à
1000°C/30min
600
500
400
300
0,0 0,2 0,4 0,6 0,8 1,0
Charge (kg)
Figure IV.21: Variation de la
micro-dureté des échantillons recuits à 1000°C /30min
en
fonction des charges appliquées pour les trois
séries.
|
Résultats et interprétations.
|
IV.3. Discussions:
La formation des carbures de tungstène et leurs
transformations avec le temps de recuit sont basées sur la
quantité de carbone contenu dans le substrat (0,7% en masse) et fourni
par diffusion thermique de ces derniers vers la couche mince de
tungstène fournit

de se réarranger pour former des carbures. La diffusion
de carbone se produit généralement à travers les
défauts de structure de tungstène, surtout par les joints des
grains et les interstices (les carbures de tungstène sont des
composés interstitiels).
S3. Ceci laisse à
la couche mince de tungstène a un effet non
négligeable sur la vitesse de formation et de croissance des carbures de
tungstène. Par ailleurs, à cause de leur rayon faible par
rapportà celui des atomes de tungstène et de fer, les atomes de
carbone pourraient diffuser plus rapidement. En plus, comme la transformation
de tungstène en carbures se fait progressivement lors de recuit
thermique, nous pouvons dire que la germination des carbures de
tungstène se produit à
e
tungstène.
-dureté de nos échantillons pour les trois
séries avec la température de recuit, la charge appliquée
et la profondeur de pénétration, épaisseur de la couche
mince de tungstène a un effet important. On remarque que la
microdureté prend des valeurs très importantes
es par les microscopies optique etélectronique à
balayage, on peut dire que, la morphologie de la surface libre
deséchantillons dépend fortement de la température et de
la durée du recuit.

Conclusion générale
Conclusion générale :
élaborées à 500°C sous vide par la
technique de pulvérisation cathodique, sur des substrats en acier de
type XC70 contenant 0.7 % en poids de carbone (d'après la norme AFNOR).
Les échantillons (couche mince de tungstène + substrat d'acier
XC70) ont subi des traitements thermiques sous vide secondaire.

morphologiques et mécaniques des couches minces
réalisées, nous avons caractérisé les
échantillons recuits et non recuits par des méthodes
variées : la diffraction de rayons X (DRX) es microscopies optique et
électronique à balayage (MEB) pour observation de la morphologie
de surface et les essais Vickers pour les mesures de la microdureté.
Nous avons, plus particulièrement, l'épaisseur de la couche mince
de tungstène, sur la réaction interfaciale.
Nous avons établi que le recuit thermique des
échantillons, indépendamment de la formation du carbure de
tungstène W2C. Ce carbure est poly - cristallin u
carbure ternaire Fe3W3 2 et 4 m.
m. En outre, nous avons remarqué la formation
2 m.
a micro-dureté augmente a
substrat. -dureté est
remarquable. En effet, les valeurs de la micro-dureté
deviennent importantes avec la diminution de l
Enfin, la morphologie de la surface libre des échantillons
dépend fortement de la température et de la durée du
recuit.
revêtements en carbure de tungstène par une
méthode indirecte qui consiste à revêtir un
diffusion du carbone et la formation de carbures.
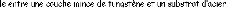
Caractérisation de la réaction interfacia

Dans ce travail, nous avons caractérisé la
réaction d e couche mince de tungséne
min, avec des substrats en acier de type XC70 (d'après
la norme AFNOR) contenant 0.7% en poids de carbone. Les échantillons
(couche mince W + substrat) ont subi des traitements thermiques sous
vide,à différentes températures (500-1000°C) et
pendant des durées variées. La la diffraction des rayons X, la
microscopie optique, la microscopieélectronique à balayage et les
essais Vickers pour mesurer la microdureté.
Les principaux résultats obtenus peuventêtre
résumés comme suit:
Pour les revêtements relativementépais (~ 6 :



Aucun

-800°C. Cependant, le recuit à une
température supérieure ou égale à 900°C
favorise la réaction entre les constituants deséchantillons (W,
Fe, C) et, par conséquent, la formation du carbure binaire W2C. L pas
été détectés.
La micro-dureté mesurée par essais Vickers,
croit légèrement avec l'élévation de la
température, surtout à partir de 900°C. Ceci est,
probablement, duà la formation et croissance du carbure W2C.
Il est montré aussi que la micro-dureté diminue
avec l'augmentation de la charge appliquée et de la profondeur de
pénétration jusqu'au substrat.
Pour les revêtements minces (2 et 4 :

Le carbure W2C se forme avec un recuità 800°C,
pendant 30 min pour tous les échantillons.
La formation du carbure ternaire Fe3W3C a lieu après un
recuit à T 900°C.
Le mono carbure WC n'est obtenu qu'avec un recuità 1000
°C pendant 60 min, seulement dans les revêtements plus minces (2
.
La morphologie de la surface deséchantillons dépend
de la température et de la durée du recuit thermique.
Mots Clés : Couches
Minces, pulvérisation cathodique RF, revêtement,
tungstène, carbures
Références :
11] E. Storms Refractory carbides , Academic Press, New
York, 1967.
12]

L.E. Toth, , Academic Press, New York, 1971.
13] Technologie des couches minces,
http://perso.wanadoo.fr.
14] Lech. Pawlowski, "dépôts physiques",
Presses Polytechniques et Universitaires Romandes.
15] Ph. Lami et Y. Pauleau, J. Electrochem.
Soc.:"Solid-State Science and Technology" 135 (1988) 980.
16] B. Grolleau, "Le vide, les couches minces", (1980)
204.
17] T. Matsushima, Thèse de doctorat, Université
d'Osaka, Japon, 1991, p. 49.
18]
Techniques de l
19] J. Lu, Z. Ye, J. H. huang, L. Wang, B. Zhao, App. Surf.
Sci., 207 (2003) 295.
110] J-A.M. DeOca-Valero, "Elaboration du carbure et nitrure
de titane par des proc édés chimiques et physiques en phase
vapeur : caract érisation de la microstructure" , Thèse de
doctorat, E.D.S.C Université Bordeaux- France (2002).
111] K.A. Taylor, Thin Solid Films, 40 (1977) 189.
112] E. Eser, R.E. Ogilvie, K.A. Taylor, J. Vac. Sci. Technol.,
15 (1978) 396.
113] Yan-Mei Jiang, "Pulvérisation cathodique assist
ée par ordinateur", Thèse de doctorat, Université de
Paris-Sud Centre d'Orsay (1992).
114] W.R. Grove, "On the electrochemical polarity ofgases "
, Philos. T. Roy. Soc. B (London),142 (1852) pp. 87 102.
115]

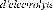
Géraldine Huertas, trode positive et
solide vitreux
sous forme de couches minces pour des micro batteries au lithium",
Thèse de doctorat, Université Bordeaux I - France (2006).
116]
Abdellah Moustaghfir, "Elaboration et
caractérisation de couches minc
de zinc. Applicationà la photoprotection
dupolycarbonate" , Thèse de doctorat, Université Blaise
Pascal France (2004).
117] De La Torre Y Ramos J, "Etudes des
propriétés optoélectroniques de structures et de
composants à base de nanostructures de Si". Thèse de
doctorat, Institut national des sciences appliquées de Lyon France
(2003).
118] S. Ido, M. Kashiwagi, M. Takahashi, Jpn. J. Appl. Phys., 38
(1999) 4450.
1191 L. Tomasini, "Les traitements de
surface sous vide", (SOLLAC, Groupe Usinor), La Revue de
Métallurgie - CIT Avril 2001.
1201 K.-S. Shim, S.-M. Kim, S.H. Bae, S.Y.
Lee, H.-S. Jung and H.-H. Park,"Fabrication And characterization of
diamond-like carbon thin films by pulsed laser deposition" , Appl. Surf.
Sci. 154-155 (2000) pp. 482-484.
1211 A.A.Voevodin and M.S.
Donley,"Preparation of amorphous diamond-like carbon by
pulsed laser deposition: a critical review ", Surf.
Coat. Technol. 82 (1996) pp.199-213. 1221 M. Bonelli, C.
Casiraghi, A. Miotello, P. Mosaner and P.M. Ossi,"Spectroscopic
characterization ofDLCfilms deposited on polycarbonate by
pulsed laser deposition" ,
Surf. Coat. Technol. 151-152 (2002) pp. 303-307.
1231 D.B. Chrisey and G.K. Hubler."Pulsed
laser deposition of thin films" . John Wiley & Sons, Inc., 1994, 613
pages. ISBN 0-471-59218-8.
1241 K. Badeker. 1907 Ann. Phys. (Leipzig)
22-749.
1251 D. Lespiaux,
nucléation/croissance et caract érisation
microstructurale
de céramiques à base SiC obtenues par CVD/CVI
dans le syst ème Si-C-H- , Thèse de doctorat : ENSPCB,
Bordeaux I France (1992).
1261 L. A. Goodman, RCA Rev., 35 (1974) 613.
1271 K. L. Chopra. S. Major, D.K. Panday,
Thin Solid Films . (1983). 102-105.
1281 S.M. Sze,"VLSI
Tecnology", 2nd eddition, McGraw-Hill International
Edditions (1988). 1291 A. Putnis and J.D.C. McConnel,
"Principles of mineral behaviour". Blackwell Sciences
Publications (1980).
1301 H.S. Carslaw and J.C. Jaeger,
"Conduction of heat insolids", Clarendon Press, Oxford
(1959).
1311 J. Cranck, "The mathematics
ofdiffusion", 2nd edition", Clarendon Press, Oxford
(1975).
1321 Y. Adda and J. Philibert, "La
diffusion dans les solides . Bibliothèque des sciences et
techniques nucléaires", tome I et tome II, Presses Universitaires
de France, Paris (1966).
1331 E. GARDES. "Diffusion du plomb dans la
monazite", Thèse de doctorat, Université Toulouse III Paul
Sabatier, France (2006).
1341 Jacques Angenault, dictionnaire
encyclopédique, 2édition, Dunod, Paris, 1995, p.71. 1351
Hicham Oudghiri, Mémoire de maître sciences (M. Sc),
Département de chimie, faculté des sciences et de génie,
université Laval, France (2000).
1361 Sergey Dudiyd ,"Microscopic Theory of
Wetting and Adhesion in Metal- Carbonitride
Systems ", Thesis for the degree of doctor of
philosophy department of Applied Physics
Chalmers university of technology göteborg university
Göteborg, Sweden (2002). 1371 J. M. Dorlot, J. P.
Baïlon, J. Masounave, "Des matériaux",
Polytechnique de Montréal, Canada (1986).
1381 Yuhuan Xu, "Ferroelectric materials and
their applications" , Elsevier, (1991). 1391
"Handbook of chemistry and physics", CRC Press, 64th edition,
1983-1984. 1401 O. Eriksson, L. Nordstrom, J. Appl. Phys., 86
(1999) pp. 3758-3767.
1411 H.W. Hugossom, O. Eriksson, Phys. Rev.B, 63
(2001) 134108.
1421 S.T. Oyama . "The chemistry of
transition metal carbides and nitrides", 1st ed., Chapman &
Hall, New York (1996) p.2.
1431 G. Aligrandi , "Matériaux
Réfractaires et Céramiques Techniques N éléments
de
Céramurgie et de Technologie", SEPTIMA, Paris
(1996) pp. 521-526.
1441 H. O. Pierson, "Handbook of refractory
carbides and nitrides, Properties, Charac-
-teristics, processing and applications" Noyes
Publications, New Jersey, U.S.A.
(1996).
1451 H. Holleck, J. Vac. Sci. Technol. A, 4
(1986) pp. 2661-2669.
1461 J. E. Sundgren, H.T.G. Huntzell, J. Vac.
Sci. Technol. A, 4 (1986) pp. 2259-2279. 1471 Jean Baralis,
Gérard Maeder, "Métallurgie, Elaboration,
structures-proprëtés, normalisation", Ed. NATHAN, Paris
(1997) p.32.
1481 B. Ghodbane, "Vieillissement de rev
êtement de carbure de tantale, sous forme de Couches minces, à
l'air et à la température ambiante", Thèse de
Magister, université de Batna (2002).
1491 Eruim Pink, Ralf Eck,"Refractory metals
and their alloys, Materials Science and Technology", Vol.10, (1994)
p.593.
1501 R. Halimi, L. Chekour, Y. Hadjar, C.
Harkati,"Etude des revêtements de substrats en acier par des films
minces de m étaux réfractaires (Ta, Ti, Cr)", Revue de
Science et Technologie, Université de constantine, 1999, p.130.
1511 J.F. Nye, "Propriétés
physiques des cristaux" , Ed. Dunod, Paris (1961).
1521 M. Zaouali, "Caractérisation par
diffraction X de l' état mécanique et microstructural
des couches minces métalliques et céramiques
obtenues par PVD et plasma", Thése de
doctorat, univ. Paris, France (1990).
1531 F.M. d'Heurle, Mat. Sci. Technol., 34
(1989) p. 53.
1541 L. Chekour, "analyse des contraintes r
ésiduelles par diffraction X dans le Zinc", Thèse de
Magister, Université de Constantine (1987).
1551 G. Meader, "Les traitements de
précontraintes des couches minces superficielles des
matériaux métalliques", Collection ENSAM,
Ed. Communications actives, Paris 61(1985).
1561 V. Teixeira, "Residual stress and
cracking in thin PVD coating", Vacuum, Vol. 64, Issues 3-4 (2002)
p.391.
1571 C. Harkati, "Elaboration et
étude des propriétés structurales et m écaniques de
revêtements durs de carbures de chrome", Thèse de Magister,
Université de Constantine (1998).
1581 E. Lassner, W.-D. Schubert,"Tungsten
properties, Chemistry, Technology of the element, Alloys, and Chemical
compounds", Kluwer Academic / Plenum Publishers, New York (1999)
pp.1-10.
1591 "Surfaces et Interfaces, indentation,
rayage et abrasion" , La Société Française de
Métallurgie et Matériaux, Colloque SF2M,
Section-Ouest: Angers 2005.
1601 Yan Boutin, L'usinage à Grande
Vitesse,
http://trametal.com, "Les
matériaux d'outils de
coupe", 2006.
1611 H.W. Hugossom, "A theoretical treatise
on the Electronic Structure ofdesigner Hard Materials", Thèse de
doctorat, Uppsala University-Suède (2001).
1621 E. Rudy, "Compendium of Phase
Diagram Data", Air Force Materials Laboratory, Wright-Patterson Air
Force Base, OH. Rep. No. AFML-TR-65-2, Part V, 1969, pp. 192 197.
1631 K. Bjorklund, "Microfabrication of
tungsten, molybdenum and tungsten carbide rods
by laser-Assisted CVD", Thèse de doctorat,
Uppsala university Suède (2001).
1641 J.P. Palmquist, "Carbide and Max-phase
engineering by thin film synthesis" , Thèse de
doctorat, Uppsala university Suède (2004).
1651 Metals Handbook, "Metallography,
Structures and Phase diagrams", Vol 8, (8 ed. ASM Handbook Committee,
USA, 1973) p.280.
1661 W.H. Rothery, R.E. Smallman,"The
Structure of metals and Alloys", 5 ed., The Metals and Metallurgy Trust
(1969) pp. 262-273.
1671 S. Ghaisas, J. Appl. Phys. 70 (1991)
7626.
1681 M. Bourahli, Thèse de Magister,
Université Farhat Abbas de sétif (1999).
1691 J.R. Tuck, A.M. Korsunsky, R.I. Davidson,
Surf. Coat. Technol., 127 (2000) pp.1-8.
[70] F. Ng-Cheng-Chin, M. Roslin, Z. H. Gu., T. Z. Fahidy, J.
Phys. D Appl. Phys., 31 (1998).
[71] L. A. Goodman, RCA Rev., 35 (1974) 613.
[72] C. M. Lampert. Sol. Energy Mater, 6-11(1981).
[73]
F.H. Lu and H.-Y. Chen, cathodic arc plasma technique on
copper substr , Surf. and Coatings technology,
130 (2000) pp.
290-296.
[74]
H. J. Shin, Y. J. Cho, J. Y. Won, H. J. Kang, C. H. Baeg, J. W.
Hong and M. Y. Wey,
ion beam assisted , Nucl. Instr. and Methods in Physics
Research B, 190
(2002) pp. 807-812.
[75]
H. H. Huang, M. H. Hon and M. C. Wang, and
characterization , Journal of Crystal Growth, 240
(2002) pp. 513-520.
[76] H.Slimani, "Elaboration et étude des
revêtements durs a base de tungst ène", Thèse de
Magister, Université de Constantine (2007).
[77] F. Khalfallah, "Elaboration et étude de carbures
de tungst ène en couches minces", Thèse de Magister,
Université de Constantine (2005).

Je dédie le présent mémoire à :
Ma mère, et mon père
et moralement,
soutien financier tout au long de ce travail, à toute ma
famille et mes amis.
A eux, je dédie ce mémoire.

Enfin, je remercie tous les ami
Le présent mémoire a été
réalisé au niveau du laboratoire des couches minces et interfaces
de la faculté des Sciences Exactes de Université
Mentouri-Constantine, sous la direction de monsieur Rachid HALIMI professeur au
Département de Physique de la
-Constantine et directeur de laboratoire des couches
minces et interfaces (LCMI), je tiens à le remercier vivement d'avoir
accepté d'être rapporteur de ce mémoire, et qu'il trouve
ici l'expression de ma très cordiale gratitude pour les conseils, le
soutien et les encouragements qu'il m'a dispensé au cours de la
réalisation de ce travail.
Mes vifs remerciements à :
Monsieur A. BOUABELLOU Professeur au Département de
Physique de la Faculté d Université Mentouri-Constantine, pour
m'avoir fait l'honneur de présider le jury de soutenance,
Monsieur S. CHEKROUD niversité
Mentouri Constantin, qui a bien voulu accepter de faire
partie du
travail,
J'adresse aussi mes remerciements à monsieurM.
BENKERRI Maître de conférence SETIF,
mon travail.
Introduction générale 01
I.1.

Chapitre I : Généralité et contexte
bibliographique.
Définition d'une couche mince 03
I.2.
Quelques techniques 04
I.2.1. Généralité 04
I.2.2. Dépôt chimique en phase vapeur (CVD).
05
I.2.3. Dépôt physique en phase vapeur (PVD)
07
I.2.3.1. Pulvérisation cathodique 07
a)-Principe de la pulvérisation cathodique DC
08
b)-Principe de la pulvérisation cathodique RF
09
c)-Pulvérisation cathodique à effet
magnétron 09
I.2.3.2. Evaporation thermique 10
I.2.3.3. Ablation laser 11
I.3. Mécanisme de formation d'une couche mince
12
I.4. Propriétés spécifiques aux couches
minces 13
I.5. Phénomène de diffusion 14
I.5.1. Les mécanismes de diffusion atomique
14
I.5.2. Les équations de diffusion 16
I.5.2.1. Première loi de Fick. 16
I.5.2.2. Deuxième loi de Fick. 17
I.5.3. Expression du coefficient de diffusion en fonction de
la température. 19
I.5.4. Les divers types de diffusion .. 20

II.1. Carbures des métaux de transition 22
II.2. Propriétés des carbures des métaux de
transition 24
II.2.1. Propriétés structurales 24
a)- Structure cristalline 24
b)- Liaisons cristallines . 27
II.2.2. Propriétés physiques et mécaniques
27

II.3. Préparat 33
II.3.1. Métallurgie des poudres 33
II.3.2. Déposition des couches minces 33
II.4. Carbures de tungstène: propriétés et
applications 34
II.4.1. Propriétés structurales. 34
II.4.2. Propriétés physiques et mécaniques.
37
II.4.3. Application des carbures de tungstène .. 38

Chapitre III : Procédures expérimentales et
méthodes de caractérisation
·
III.1. Préparation des échantillons 39
III.1.1. Préparation des substrats 39
a)- Polissage mécanique 40
b)- Nettoyage chimique et ionique 40
III.1.2. Préparation du dépôt de
tungstène 41
III.1.3. Les traitements thermiques appliqués (recuits)
42
III.2. Les méthodes de caractérisation 43
III.2.1. La diffraction des rayons X (DRX) 43
III.2.1.1 Le Principe 44
III.2.1.2 Les conditions expérimentales 45
III.2.1.3 Détermination de la taille de grain 46
III.2.2. La microscopie électronique à balayage
(MEB) 47
III.2.2.1 Le principe 47
III.2.2.2 Les conditions expérimentales 49
III.2.3. Le microscope optique 49
III.2.3.1 Le principe 49
III.2.3.2 Les conditions expérimentales 50
III.2.4. La micro-dureté 50

III.2.4.1 Princi 50
III.2.4.2 Profondeur de pénétration de l'indenteur
dans un essai d'indentation 52
III.2.4.3 Les conditions expérimentales 52
Chapitre IV : résultats expérimentaux et
interprétation.
IV.1. Caractérisation des couches minces de la
série S3. 53
IV.1.1. Etudes structurale et morphologique 53
IV.1.1.1 Analyse des échantillons non recuits et recuits
entre 600 et 700°C 53
IV.1.1.2. Analyse deséchantillons recuits entre 900 et
1000°C 56
IV.1.1.3. Evolution de la structure de la couche de
tungstène avec la
température de recuit
57
IV.1.2. Etudes microstructurale et morphologique . 59
a)- Observation par microscope optique 59
b)- Observation par microscopie électronique à
balayage (MEB) 61
IV.1.3 Mesures de la micro-dureté 63
a)- Variation de la micro-dureté en fonction de la
température 63
b)- Variation de la micro-dureté en fonction de la charge
appliquée 64
c)- Variation de la micro-dureté en fonction de la
profondeur de pénétration ... 65
IV.2. Etude comparative entre la série S3 et les deux
autres séries S1 et S2 66
IV.2.1. Études structurale et morphologique 66
IV.2.2. Études microstructurale et morphologique 70
IV.2.3. Etude de la micro-dureté 71
IV.3. Discussion 73
Conclusion générale . 74
Références ... 75
Annexe .. 80
| 

