GENERALITES
1. GENERALITES SUR L'INFECTION à VHB
1.1. HISTORIQUE
En 1964, Baruch Samuel BLUMBERG, médecin et biochimiste
américain, travaillant pour le National Institute of Health,
s'intéresse à la variabilité antigénique entre les
individus et au sein des différentes populations. Il émet
l'hypothèse selon laquelle des patients ayant reçu un grand
nombre de transfusions sanguines doivent avoir développé des
anticorps contre les antigènes qu'ils ne possèdent pas. Il met en
présence des échantillons de sang de patients
polytransfusés avec des sérums de personnes indemnes de toute
transfusion. Il observe alors, en immuno-diffusion, une ligne de
précipitation pour chaque système antigène-anticorps
révélé.
Ensuite, il remarque qu'un échantillon sanguin d'un
patient hémophile polytransfusé présente la
caractéristique de former une ligne de précipitation originale
avec un seul sérum, celui d'un Aborigène australien. Ce
sérum contient donc un antigène qui n'existe pas dans les autres
lots ; BLUMBERG le baptise : « Antigène
Australia ». Ses travaux consistent alors à établir la
répartition de cet antigène dans diverses populations : un
sérum sur 1000 est positif en Amérique du Nord contre 15
sérums sur 100 dans certaines îles du Pacifique ; il existe
donc une variabilité dans la distribution de cet antigène. Reste
à trouver l'origine de ce portage antigénique.
En 1966, le changement de statut sérologique d'un
patient initialement dépourvu d'antigène Australia renforce
l'hypothèse d'une infection par un agent viral et ce patient a
présenté une hépatite pendant la période de
séroconversion. Ceci conduit à tester de nombreux
échantillons de sang de patients aux antécédents
d'hépatite. A la fin de l'année 1966, la preuve est faite que le
portage de l'Antigène Australia est lié à une
hépatite virale. BLUMBERG établit un protocole de
dépistage du sang destiné aux transfusions, éliminant tous
les lots porteurs de l'antigène; rapidement une nette diminution du
nombre d'hépatite post-transfusionnelle est constatée [12].
L'observation au microscope électronique du
sérum contenant l'Antigène Australia révèle la
présence de particules de 42 nanomètres de diamètre dont
l'antigène Australia constitue une partie. La structure de ce virus
aujourd'hui appelé VHB est vite élucidée.
L'antigène Australia est aujourd'hui connu sous le nom d'antigène
de surface du VHB (AgHBs).
Par la suite, des découvertes provenant du monde entier
n'ont cessé d'accroitre les connaissances sur le virus, notamment depuis
l'avènement de la biologie moléculaire :
· En 1971, DANE découvre la particule qui porte
son nom, d'un diamètre de 40 à 42 nm et qui correspond au virion.
Il apparait sous la forme de petite sphère ou de petit tube
correspondant à des fragments de l'enveloppe du virus
lui-même ;
· En 1972, MAGNIUS découvre l'antigène HBe
soluble qui est le témoin de la multiplication virale.
1.2. CARACTERISTIQUES VIROLOGIQUES
Le VHB appartient à la famille des hepadnaviridae, avec
le virus de l'hépatite de la marmotte, le virus de l'hépatite du
canard et quelques autres variantes aviaires et mammifères. Tous les
virus de la famille sont hépatotropes et ont le même cycle de
réplication chez l'hôte [13].
1.2.1. ULTRASTUCTURE
L'observation en microscopie électronique de
sérum infecté par le VHB met en évidence trois types de
particules (Figure1) correspondant aux différentes formes virales
[14] :
· La particule de DANE ou virion complet, sphère
de 42 nm de diamètre, constituée d'une enveloppe entourant la
capside virale, à l'intérieur de laquelle se trouvent la
molécule d'ADN viral et 2 enzymes (une ADN polymérase et une
protéine kinase) : c'est la particule infectante du VHB ;
· Des particules de forme sphérique de 17 à
25 nm de diamètre ;
· Des particules filamenteuses de longueurs variables,
non infectantes, correspondant à des protéines d'enveloppe
synthétisées en excès.
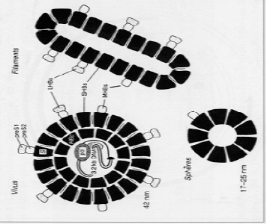
Figure 1. Ultra structure du virus de
l'hépatite B en représentation schématique [14]
1.2.2. ORGANISATION GENOMIQUE
Le génome du VHB est une molécule circulaire
bicaténaire sur les trois quarts du cercle et monocaténaire sur
un quart, constitué d'environ 3200 paires de bases (Figure 2) [15].
On identifie :
· Un brin négatif de 3200 bases, maintenu sous
forme circulaire par son extrémité 5' où se fixe l'ADN
polymérase virale ;
· Un brin positif complémentaire de longueur
variable.
L'organisation génomique de cette molécule d'ADN
est compacte. Elle comprend quatre (4) cadres de lecture ou régions se
chevauchant, permettant la transcription et la traduction des gènes
viraux, pour aboutir à la synthèse de 7 protéines
différentes. Il s'agit de :
· La région
pré-S/S, codant pour trois protéines de
surface : la protéine L (Large), la protéine M (Middle) et
la protéine S (Small). Celle-ci détermine
l'antigénicité HBs.
Cette région est organisée en une région
S, précédée en amont par une
région pré- S2,
elle-même précédée d'une région
pré-S1. A chaque région
correspond un codon permettant la lecture des 3 gènes [15] :
Ø La protéine L correspond à l'expression
du gène pré-S1 +
pré- S2+S ;
Ø La protéine M à l'expression du
gène pré-
S2+S ;
Ø La protéine S à l'expression du
gène S.
· La région pré-C/C,
codant pour 2 protéines : l'antigène HBe, un
peptide de 25 kDa qui, après maturation dans les membranes du
réticulum endoplasmique de l'hôte, aboutit à la
sécrétion d'un peptide soluble de 15 kDa dans le plasma des
patients infectés et l'antigène HBc, protéine
cytoplasmique de 21 kDa encore appelée protéine de core,
détectable dans les hépatocytes infectés mais non
sécrétée dans le plasma.
· La région P, codant
pour l'ADN polymérase, une enzyme permettant la synthèse d'ADN
viral. Cette région est formée de 3 domaines fonctionnels et d'un
domaine non fonctionnel dans l'ordre suivant :
Ø Un domaine N-terminal, lié à la partie
5' du brin négatif de l'ADN viral, il sert également d'amorce
à l'initiation de la synthèse de ce brin négatif par son
activité primase ;
Ø Un domaine intermédiaire non essentiel,
espaceur (spacer), dont la taille et le repliement
permettent l'interaction des différents domaines avec le
génome ;
Ø Un domaine pour la transcriptase
inverse/ADN polymérase ;
Ø Un domaine pour la
RNaseH.
· La région X, codant pour la protéine X
qui a un rôle important dans la transactivation de la transcription
virale (augmentation des ARN messagers) et dans l'augmentation de
l'activité de gènes de la croissance cellulaire(c-myc et c-fos),
contribuant ainsi au processus d'oncogenèse de la cellule
infectée [17].
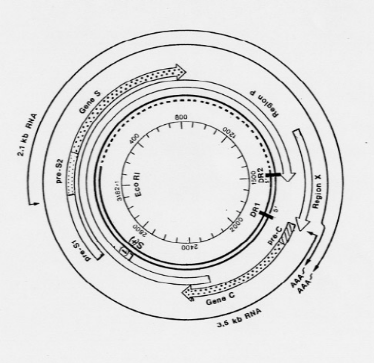
Figure 2. Organisation génomique du VHB
[15]
1.2.3. REPLICATION VIRALE
Il n'existe pas de modèle cellulaire permettant la
culture virale, ce qui complique la compréhension du cycle viral dans la
cellule humaine. Seuls certains primates, dont le chimpanzé, constituent
des modèles de choix pour l'étude du VHB.
Le VHB a un tropisme essentiellement hépatocytaire,
mais l'ADN viral peut être retrouvé dans les cellules de la
moelle osseuse, les cellules mononucléées du sang
périphérique (lymphocytes B et T, monocytes), les cellules
pancréatiques, rénales et cutanées. Cependant les formes
réplicatives sont exceptionnelles en dehors des hépatocytes.
Le cycle viral débute par l'entrée dans la
cellule. Après décapsidation cytoplasmique, le génome
viral pénètre dans le noyau cellulaire. Le brin positif est
complété, donnant naissance à un ADN bi caténaire
circulaire refermé sous forme super enroulée
(supercoiled). C'est le ccDNA
(covalently closed circular DNA).
La transcription s'initie dans le noyau à partir du
brin négatif, produisant un ARN prégénomique de 3,5 kb et
des ARN messagers subgénomiques de 2,4 à 2,1 kb et 0,5 kb, qui
codent pour les protéines de surface, de la capside, mais aussi pour la
protéine transactivatrice X et l'ADN polymérase.
Après l'encapsidation de l'ARN
prégénomique dans le cytoplasme, la transcriptase inverse virale
produit un brin d'ADN négatif, qui sert de matrice pour la
synthèse partielle du brin positif, alors que l'activité RNaseH
virale dégrade l'ARN prégénomique. Le virus finit sa
maturation dans le réticulum endoplasmatique cellulaire par acquisition
de son enveloppe, puis quitte la cellule par un phénomène de
bourgeonnement membranaire [17].
La synthèse des ccDNA à l'intérieur du
noyau cellulaire joue un rôle important dans l'évolution de
l'infection. En effet, cette forme génomique extrêmement stable,
parfois qualifié de mini chromosome, persiste sous forme
épisomale au sein de la cellule hépatique et probablement au sein
d'autres cellules permissives. Cela est à l'origine du portage chronique
du VHB, des phénomènes de réactivation et explique que
l'on puisse détecter l'ADN viral après disparition de l'AgHBs
sérique [17].
L'implication de la transcriptase inverse dans le cycle
réplicatif est à l'origine des mutations dont le nombre est plus
élevé que celle rencontrées dans la réplication des
virus à ADN classiques et explique l'apparition de variants du VHB. En
effet la demi-vie moyenne du VHB dans le sang est de 1 à3 jours et le
taux de production des virions serait proche de 10-11 par jour ; d'autre
part, la transcriptase inverse aurait un taux d'erreur estimé à
10-` par base et par cycle, sans système de correction des erreurs.
Cette combinaison d'une réplication quotidienne élevée et
d'un nombre important d'erreurs non corrigées explique la survenue des
variants génétiques du VHB [18].
1.3. EPIDEMIOLOGIE
1.3.1. Dans le monde
L'hépatite B est une maladie ubiquitaire. On estime que
2 milliards de personnes dans le monde ont eu un contact avec le VHB et environ
360 millions ont développé une infection chronique. Celles-ci ont
un risque accru de développer une cirrhose hépatique, puis un
carcinome hépatocellulaire [19].
La prévalence de l'infection et le mode de transmission
varient en fonction des régions du globe (Figure 3). On
distingue :
o Les régions de forte
endémicité, définies par une prévalence de
l'infection virale chronique supérieure à 8%. Il s'agit de
l'Afrique subsaharienne et des pays asiatiques. La contamination est
essentiellement périnatale à partir d'une mère
infectée ou survient tôt dans l'enfance ; or l'infection de
l'enfant devient plus volontiers chronique, expliquant la forte
prévalence dans ces régions.
o Les régions d'endémicité
intermédiaire ont une prévalence de l'infection
chronique à VHB comprise entre 8 et 1% ; il s'agit des pays
méditerranéens et des pays de l'Europe de l'Est. La contamination
est familiale, sexuelle, périnatale et nosocomiale.
o Les régions de faible
endémicité ont une prévalence de l'infection
chronique à VHB inférieure à 1% ; il s'agit de
l'Europe du Nord, de l'Ouest, de l'Amérique du Nord et de l'Australie.
La transmission se fait essentiellement par voie sexuelle ou par échange
d'aiguilles contaminées chez les utilisateurs de drogues.

Figure 3. Répartition de la
Prévalence de l'infection à VHB dans le monde [19]
1.3.2. MODE DE TRANSMISSION
Le VHB est très contagieux, environ 100 fois plus que
le VIH et 10 fois plus que le VHC [17]. Le réservoir viral est humain et
la transmission inter humaine. On distingue essentiellement quatre modes de
transmission.
1.3.2.1. Transmission sexuelle
Elle est fréquente partout dans le monde, mais c'est un
mode important de transmission dans les zones de faible endémie.
1.3.2.2. Transmission parentérale
Elle résulte de l'injection ou de contact avec des
produits sanguins ou des dérivés sanguins infectés, de
l'utilisation de matériel médico-chirurgical souillé
(chirurgie, hémodialyse, odontologie, acupuncture et
mésothérapie), de toxicomanie intraveineuse, les tatouages et le
piercing [23].
1.3.2.3. Transmission mère -enfant
La transmission survient chez les femmes enceintes
présentant une hépatite aigue au deuxième et surtout au
troisième trimestre de la grossesse et chez les porteuses chroniques du
virus. Pour ces dernières, le risque de transmission est faible (environ
20% en dehors de tout traitement) chez les porteuses de l'AgHBs sans
réplication virale détectable dans le sérum. A l'inverse,
il est élevé (de l'ordre de 80%) chez les porteuses chroniques
présentant les marqueurs de réplication virale [23].
Dans tous les cas, la transmission est périnatale soit
lors de l'accouchement par contact avec les sécrétions
maternelles infectées dans la filière génitale, soit dans
les mois suivant l'accouchement par contact avec les sécrétions
maternelles infectées (lait, sueur, larmes).
Ce mode de contamination est présent dans le monde
entier, mais prédomine dans les régions de forte
endémicité [23].
1.3.2.4. Transmission interindividuelle
directe
Elle se fait par contact direct interindividuel. Elle semble
particulièrement fréquente en Afrique sub-saharienne où le
contage a souvent lieu entre les enfants en bas âge à la maison
familiale, dans les crèches ou à l'école. Les vecteurs de
la transmission sont alors de très petites quantités de sang ou
de salive à la faveur d'excoriations cutanées ou muqueuses
[23].
1.4. PHYSIOPATHOLOGIE
La physiopathologie de l'infection à VHB est complexe.
En effet, la réplication virale n'est pas directement
cytopathogène pour l'hépatocyte, mais c'est la réaction de
l'hôte vis-à-vis du virus qui est déterminante dans la
physiopathogénie. Il s'agit d'une réponse à la fois
humorale et cellulaire, responsable des lésions hépatiques et des
symptômes [16].
La réponse humorale est fondée sur les
propriétés des récepteurs d'immunoglobulines des cellules,
qui reconnaissent les antigènes viraux à la surface des
hépatocytes infectés ou sous leurs formes solubles dans le
sérum. La réponse cellulaire fait intervenir les lymphocytes T et
les cellules présentatrices d'antigène, essentiellement les
macrophages.
A la surface des hépatocytes infectés, les
molécules HLA de classe 1 présentent des fragments
antigéniques, le plus souvent l'AgHBc, métabolisé dans le
cytosol de l'hépatocyte. Le couple HLA de classe 1-AgHBc est reconnu par
les lymphocytes T CD8 cytotoxiques via un récepteur spécifique.
Ceci induit un processus de lyse cellulaire médié par la
protéine Fas, des cytokines et des perforines.
La capacité des molécules HLA de classe 1 à
présenter l'antigène dépend de la variabilité du
Complexe Majeur d'Histocompatibilité (CMH) et du répertoire des
récepteurs des lymphocytes T de l'hôte. L'intensité de la
réponse va donc varier d'un individu à l'autre [13].
Dans les tissus, les macrophages vont phagocyter les virions
libres circulants. Après protéolyse dans leurs compartiments
d'endocytose, les antigènes viraux sont associés aux
molécules HLA de casse 2 et présentés aux lymphocytes T
CD4+Helpers. La résultante en est une augmentation de la synthèse
de cytokines activatrices de la prolifération des lymphocytes T et une
augmentation de la présentation antigénique par les
molécules HLA de classe 1 à la surface des hépatocytes,
tendant à la clairance virale.
La nature et la qualité de la réponse immune
obéit à un déterminisme multifactoriel, notamment
génétique et aboutit à quatre types de relation
hôte-virus [13]:
Ø La réaction immune de l'hôte est forte,
aboutissant à l'élimination des virus circulants et des
hépatocytes infectés ; c'est l'hépatite aigue
guérie. Dans l'hépatite aigue fulminante, cette réaction
est suraiguë, aboutissant à une nécrose
hépatocellulaire massive ;
Ø La réaction immune de l'hôte est faible
mais adéquate. L'infection reste asymptomatique et évolue vers la
guérison ;
Ø La réaction de l'hôte est faible et
inadéquate. Il s'installe une tolérance partielle combinant la
réplication virale prolongée (AgHBs persistant) et une
destruction tissulaire hépatique à bas bruit. Cette situation
d'hépatite chronique peut se prolonger plusieurs mois, voire des
années et aboutir à la cirrhose. Au cours de cette période
et sous la dépendance de cofacteurs alimentaires et toxiques, peut se
produire la transformation hépatocellulaire conduisant au cancer
primitif du foie ;
Ø La réaction immune de l'hôte est nulle.
Il existe une tolérance totale à la réplication virale.
C'est la situation du portage chronique asymptomatique ou portage inactif.
1.5. CLINIQUE
On distingue l'hépatite virale aiguë et
l'hépatite virale chronique. Dans les deux cas, l'infection peux
être symptomatique ou non.
1.5.1. Incubation
La durée d'incubation est de 50 à 100 jours, 10
semaines en moyenne. Dans 90% des cas l'infection reste asymptomatique d'autant
plus que le sujet est jeune.
1.5.2. INFECTION AIGUË PAR LE VHB
[13]
1.5.2.1. Forme classique
La forme classique de l'infection aiguë à VHB est
la forme ictérique, observée dans 10% des cas. Elle
évolue en 3 phases :
Ø La phase pré-ictérique
qui dure 3 à 7 jours, elle est absente dans 20% des cas. Elle est
caractérisée par des signes non spécifiques :
céphalées, asthénie, anorexie, fièvre, plus
rarement les arthralgies, myalgies, nausées, pesanteur de l'hypochondre
droit, foie sensible à la palpation et rash cutané ;
Ø La phase ictérique qui dure 2
à 6 semaines, caractérisée par un ictère
cutanéo-muqueux, rarement accompagné de prurit. Lorsque
l'ictère apparait la fièvre disparait. Les urines sont brun
acajou, les selles incomplètement décolorées.
L'asthénie est constante et dure tout au long de la phase
ictérique ;
Ø La phase de convalescence voit la
disparition de l'ictère et des signes généraux
Biologiquement la cytolyse est l'élément
primordial avec des taux supérieurs à dix fois la normale,
prédominant sur l'Alanine Amino-Transférase (ALAT). Il n'y a pas
d'insuffisance hépatocellulaire. Le taux de prothrombine(TP) reste
supérieur à 60%, sauf dans les formes sévères
(TP< 50%), imposant une hospitalisation pour la surveillance. Il existe une
cholestase avec élévation de la bilirubine totale et surtout la
fraction conjuguée. Enfin, les marqueurs de l'inflammation sont
perturbés avec élévation de la vitesse de
sédimentation et des bêta et gamma globulines.
1.5.2.2. Forme fulminante
Dans un cas sur 1000, l'hépatite est suraiguë.
Elle met en jeu le pronostic vital car en l'absence de la transplantation
hépatique en urgence, la mortalité est d'environ 90%. Les signes
d'alerte à la phase initiale sont une encéphalopathie
hépatique caractérisée par une inversion du rythme
nycthéméral, un astérixis et un syndrome confusionnel,
associés à une diminution du TP (TP< 30%) et du facteur V,
ayant comme conséquence des hémorragies cutanéo-
muqueuses. La cytolyse est très importante et il existe une
hypoglycémie en rapport à une insuffisance
hépatocellulaire.
Lorsqu'il y a une évolution favorable, le passage
à la chronicité est exceptionnel [19].
1.5.3. INFECTION CHRONIQUE PAR LE VHB
L'infection chronique par le VHB est définie par le
portage pendant plus de six mois de l'AgHBs. Elle survient chez 5 à 10%
des adultes infectés immunocompétents, plus fréquemment
chez les immunodéprimés et chez 90% des nouveaux nés
infectés.
Elle est caractérisée par un polymorphisme,
incluant les patients atteints d'hépatite chronique et les porteurs
inactifs de l'AgHBs.
1.5.3.1. Hépatite chronique
Elle concerne environ deux tiers des porteurs de l'AgHBs. Elle
est définie par l'association du portage chronique de l'AgHBs et de la
présence des lésions hépatiques notamment la
nécrose hépatocytaire, l'inflammation et la fibrose [24].
Sur le plan clinique, elle est généralement
asymptomatique et découverte à l'occasion d'un bilan
systématique, parfois même au stade de cirrhose. Lorsque les
signes cliniques sont présents, ils sont peu évocateurs :
asthénie, anorexie, gène sous costale et plus rarement un prurit
et un ictère dans les formes choléstatiques [23].
Sur le plan biologique, une cytolyse est le plus souvent
retrouvée mais moins importante que dans les formes aiguës (entre
une et cinq fois la normale), prédominant sur les ALAT. Les autres
marqueurs hépatiques sont normaux en dehors des formes
choléstatiques. Il peut exister un syndrome inflammatoire avec
élévation des immunoglobulines prédominant sur les IgG
[23].
Sur le plan histologique, une biopsie hépatique permet
de poser le diagnostic de certitude. Elle renseigne sur 3
éléments fondamentaux :
Ø L'activité hépatique avec des
lésions de nécrose et d'inflammation portales,
péri-portales et lobulaires ;
Ø La fibrose en fonction des lésions
cicatricielles, désorganisant progressivement la structure
parenchymateuse, jusqu'à aboutir à la cirrhose;
Ø Les lésions éventuellement
associées comme la stéatose, la surcharge en fer ou des
lésions d'hépatite alcoolique.
L'infiltrat inflammatoire est souvent intense,
constitué de cellules mononuclées, typiquement lymphocytaires,
les cellules CD4+ sont plus volontiers présentes dans les espaces
portes, alors que les CD8+ prédominent au niveau parenchymateux, dans
les zones de nécrose.
L'évaluation de l'activité cellulaire et de la
fibrose se fait au moyen de scores histologiques tels que le score de KNODELL
ou le score de METAVIR, plus récent et mieux reproductible (Tableau
I).
Tableau I : le score de METAVIR
|
STADE DE FIBROSE
GRADE D'ACTIVITE (nécrose)
|
|
F0 : Pas de fibrose
A0 : Pas d'activité
F1 : Fibrose portale sans septa
A1 : Activité minime
F2 : Fibrose portale et quelques septa
A2 : Activité
modérée
F3 : Fibrose septale sans cirrhose
A3 : Activité
sévère
F4 : Fibrose septale avec cirrhose
|
Il est à souligner que deux méthodes
récentes non invasives permettent d'évaluer la fibrose
hépatique. Il s'agit de :
· Dosage des marqueurs biochimiques des maladies
hépatiques, Fibrotest et Actitest, permettant des estimations de la
fibrose et de l'activité nécrotico- inflammatoire en fonction du
dosage de 5 marqueurs hépatiques : alpha 2 macroglobuline,
haptoglobine, apolipoprotéine A1, bilirubine totale et gamma GT [23].
· Fibroscan qui estime la fibrose hépatique par
mesure de l'élasticité du foie (kPA) en utilisant une nouvelle
technique qui est l'élastométrie impulsionnelle. Les
résultats sont exprimés en kPA avec les valeurs limites
suivantes : 7,5 kPA correspondent à F=2.
1.5.3.2. Hépatite chronique AgHBe
négatif
La séroconversion dans le système HBe marque
classiquement la transition vers la phase de latence, mais dans 1 à 5%
des cas persistent les activités biologique et histologique avec un haut
niveau de réplication virale. Cette situation est due à deux
types de mutation :
Ø Mutants pré-core, qui ont une substitution de
la guanosine en position 1896 par une adénosine (G1896A) qui crée
un codon stop en position 28. Cette mutation entraine un arrêt de
l'expression de l'AgHBe ;
Ø Mutant Basal Core Promoteur (mutant BCP),
présentant une double substitution au niveau du gène X, avec
remplacement de l'adénosine en position 1762 par une thymidine et de la
guanosine en position 1764 par une adénosine (A1762T/G1764A). cette
double mutation entraine une réduction de 70% de la
sécrétion de l'AgHBe.
Ces variants viraux coexistent initialement avec les souches
sauvages, qui perdent progressivement leur avantage sélectif aux
dépend des souches virales mutées émergentes [1].
1.5.3.3. Portage chronique inactif de l'AgHBs
[24]
Le portage chronique inactif de l'AgHBs associe : la
présence pendant plus de 6 mois de l'AgHBs ; l'absence de signes
cliniques ; l'absence d'anomalies biologiques ; l'absence d'infection
par le VHD ou le VHC ; la présence d'anticorps anti-HBe et la
charge virale inférieure à 105 copies/ml.
Cette définition regroupe des patients dont l'infection
n'est pas active mais sans préjuger de l'évolutivité
antérieure et des éventuels retentissements hépatiques
qu'elle aurait pu causer.
1.6. COMPLICATIONS
1.6.1. Cirrhose
La cirrhose est un événement crucial dans
l'histoire de l'infection à VHB, car ses complications propres, de
l'hypertension portale et de l'insuffisance hépatocellulaire sont en
grande partie responsables de la morbidité et de la mortalité de
cette infection.
L'incidence annuelle de la cirrhose chez les patients atteints
d'hépatite chronique AgHBe positif est de 2 à 5,5% et de 8
à 10% chez les patients avec une hépatite chronique AgHBe
négatif [1].
1.6.2. Carcinome hépatocellulaire
La fréquence annuelle de CHC varie en fonction des
populations : chez les porteurs chroniques sans cirrhose, le taux annuel
est inférieur à 0,2% dans les pays occidentaux contre 0,6% en
Asie et l'Afrique. Chez les cirrhotiques ce chiffre s'élève
à 2% [1].
Les patients atteints d'une hépatite chronique B
à AgHBe négatif, mutant BCP ont un risque accru de
développer un CHC par rapport à l'ensemble des porteurs
chroniques de l'AgHBs [24].
1.6.3. Manifestations extra-hépatiques
associées à l'hépatite chronique B
L'hépatite chronique B peut s'accompagner des
manifestations extra-hépatiques liées à la formation des
complexes immuns [23] :
· La périarthrite noueuse, observée chez 1
à 2% des porteurs chroniques du VHB. Elle est due à la
présence de complexe AgHBs-anticorps anti-HBs circulants et une
diminution du complément sérique [23] ;
· La glomérulonéphrite
membrano-proliférative, dont le diagnostic se fait par la mise en
évidence en immunofluorescence de l'AgHBs au sein des
dépôts glomérulaires de complexes immuns [23].
1.7. DIAGNOSTIC
Le diagnostic définitif de l'infection à VHB
repose sur l'utilisation des marqueurs sérologiques, associée
à l'étude des marqueurs de réplication du VHB et aux
stades histologiques hépatiques.
1.7.1. MARQUEURS SEROLOGIQUES
Ø Le système HBs : l'AgHBs est le marqueur
sérologique nécessaire à tout diagnostic d'infection par
le VHB. Il apparait dans le sang pendant la phase d'incubation, 1 à 6
semaines avant les signes cliniques ou biochimiques. Il disparait pendant la
phase de convalescence des hépatites aigues qui guérissent. Sa
disparition signe l'évolution favorable et sa persistance pendant plus
de 6 mois définit le passage à la chronicité.
La présence de l'anticorps anti-HBs permet d'affirmer
la guérison de l'hépatite aiguë B ; il apparait en
général 2 à 8 semaines après la disparition de
l'AgHBs et le plus souvent après amendement des signes cliniques.
L'anticorps anti-HBs persiste au moins 10 ans. C'est un anticorps neutralisant
dont la présence permet d'affirmer l'efficacité d'un vaccin
[23].
Ø Le système HBc : la recherche de l'AgHBc
ne se fait pas en routine clinique. En effet, il est présent à la
surface des hépatocytes infectés où il est la cible de la
réponse immunitaire, responsable de la destruction cellulaire. Il est
détectable en immuno-histochimie à des fins
expérimentales. Par contre l'anticorps anti-HBc dirigé contre la
capside du VHB est le marqueur de choix pour témoigner d'un contact avec
le VHB. En effet, on le retrouve à la fois dans les infections actives
et guéries.
Les IgM anti-HBc apparaissent 1 à 2 semaines
après l'apparition de l'AgHBs, signant la primo-infection et peuvent
persister plusieurs mois. Puis apparaissent les IgG anti-HBc, que l'infection
ait été aiguë et guérie ou qu'elle ait
évolué vers la chronicité. Ceux-ci persistent quasiment
à vie [23].
L'anticorps anti-HBc est un meilleur marqueur
sérologique d'infection ancienne que l'anticorps anti-HBs, car il n'est
pas produit par la vaccination. Il est présent lors de la fenêtre
sérologique où il y a absence de l'AgHBs et de l'anticorps
anti-HBs.
Ø Le système HBe : l'AgHBe est
sécrété sous forme soluble dans le sang. Sa
présence signe une réplication active du VHB. Elle est
généralement parallèle à la présence d'ADN
viral dans le sang. Cette présence est un élément
important en faveur de la contagiosité du patient.
La disparition de l'AgHBe est plus précoce que celle de
l'AgHBs. Associée à l'apparition d'anticorps anti-HBe, elle
définit la séroconversion dans le système HBe. Cette
séroconversion n'est pas un signe formel de guérison, mais un
élément pronostique favorable, généralement
associé à l'arrêt de la réplication virale. Dans 1
à 2% des cas de séroconversion dans le système HBe, l'ADN
viral reste détectable dans le sérum, définissant le
groupe des hépatites B chroniques à AgHBe négatif [23].
1.7.1.1. INTERPRETATION
Les figures 4 et 5 illustrent la cinétique des
marqueurs virologiques dans le sérum au cours de l'hépatite B
aiguë et chronique, alors que le tableau 2 récapitule l'ensemble
des situations sérologiques qu'il est possible de rencontrer au cours de
l'infection chronique par le VHB.

Figure 4. Cinétique de marqueurs
sériques de l'Hépatite virale B aiguë [17]

Figure 5. Cinétique des marqueurs
sériques au cours d'une hépatite virale B chronique [17]
|
ANTIGÈNES
|
ANTICORPS
|
DNA
|
|
Ag HBs
|
Ag HBe
|
Ac anti HBs
|
Ac anti HBc
|
Ac anti HBe
|
DNA du virus
|
|
Hépatite aiguë au début
|
+
|
+
|
-
|
-
|
-
|
+
|
|
Hépatite aiguë phase d'état
|
+
|
+
|
-
|
+ (IgM)
|
-
|
+
|
|
Hépatite aiguë phase post-ictérique
|
V
|
-
|
V
|
+ (IgM)
|
+
|
V
|
|
Guérison
|
-
|
-
|
+
|
+ (IgM)
|
+
|
-
|
|
Hépatite chronique avec virus circulant
|
+
|
+
|
-
|
+
|
-
|
+
|
|
Hépatite chronique sans virus circulant
|
+
|
-
|
-
|
+
|
+
|
-
|
|
Porteur asymptomatique avec virus circulant
|
+
|
+
|
-
|
+
|
-
|
+
|
|
Porteur asymptomatique sans virus circulant
|
+
|
-
|
-
|
+
|
+
|
-
|
Tableau II : Différentes
situations sérologiques rencontrées au cours de l'infection
à
VHB
1.7.2. MARQUEURS DE REPLICATION VIRALE
La recherche de l'ADN viral dans le sang est la méthode
de référence pour détecter la présence de virion.
Il existe :
Ø des techniques basées sur le principe
d'hybridation de l'ADN avec amplification du signal (bDNA). Les
résultats sont exprimés de façon quantitative. La limite
est le manque de standardisation des kits de dosage et de l'unité de
mesure de l'ADN du VHB. Les tests ont des sensibilités et des gammes de
linéarité différentes ;
Ø des techniques basées sur le principe de la
PCR (Polymerase Chain Reaction) classique ou en temps réel. La
sensibilité est meilleure avec des seuils de détection à
200 copies par ml, voire à 64 copies par ml pour les techniques de PCR
en temps réel (Cobas Taqman).
Il y a actuellement très peu de données pour
trouver une signification clinique aux différents niveaux des charges
virales. Cependant, de nombreuses études anciennes laissent penser que
le niveau de 105 copies/ml, seuil de sensibilité des techniques
n'utilisant pas la PCR, représente le seuil au dessous duquel
l'hépatite serait non progressive et inactive [1].
1.7.3. HISTOLOGIE
La Ponction Biopsie Hépatique (PBH) permet de confirmer
le diagnostic d'hépatite chronique B, de détecter les autres
causes de maladie hépatique, de juger la sévérité
de l'activité nécrotico-inflammatoire, ainsi que de la fibrose
[1].
L'infection par le VHB peut être associée
à une maladie du foie active ou inactive. Une maladie active secondaire
à l'infection par le VHB se définit par un taux de transaminases
élevé et/ou une inflammation à l'histologie
hépatique qui ne peut être expliquées par une autre cause
que l'infection par le VHB. Une maladie inactive du foie est définie par
un taux de transaminases normal et ou l'absence ou une minime inflammation
à l'histologie [1].
1.8. TRAITEMENT
1.8.1. TRAITEMENT CURATIF
1.8.1.1. Principes généraux
[23]
A la phase aiguë de l'hépatite virale B, le
traitement antiviral spécifique est inutile. Seules des mesures
symptomatiques peuvent être prises, associées à
l'éviction de l'alcool et des médicaments
métabolisés par le foie. Une transplantation hépatique
d'urgence est nécessaire dans la forme fulminante.
Le traitement antiviral spécifique trouve sa place dans
l'hépatite chronique B, l'objectif étant d'obtenir l'arrêt
de la réplication virale, afin de prévenir l'évolution
naturelle de la maladie vers les complications.
La réponse au traitement comporte 3 phases :
Ø La première phase marquée par une
diminution de la réplication virale, traduite par une diminution de
l'ADN viral sérique. L'activité de l'hépatite chronique
régresse, la fibrose se stabilise et peut même diminuer ;
Ø La deuxième phase intervient lorsque
l'activité antivirale est suffisamment forte et prolongée,
accompagnée d'une réponse immunitaire adaptée avec la
clairance des hépatocytes infectés. Une séroconversion HBe
peut intervenir et le risque de réactivation est faible ;
Ø La troisième phase marquée par une
réplication virale complètement interrompue (l'ADN
indétectable). La séroconversion HBe est stable, l'AgHBs
disparait avec ou sans apparition des anticorps anti-HBs. Le risque de
réactivation spontanée est nul et l'activité disparait.
1.8.1.2. Antiviraux actuellement disponibles
[23]
Actuellement en France, trois molécules ont
l'autorisation de mise sur le marché dans le traitement de
l'hépatite virale chronique B. il s'agit de l'interféron, de la
lamivudine et de l'adénofovir.
1.8.1.2.1. Interféron
Les interférons sont des glycoprotéines de la
famille des cytokines endogènes sécrétées par les
lymphocytes et les macrophages activés, en réponse à de
nombreux stimuli en particulier les infections virales. Il en existe deux types
d'activités biologiques différentes. Ce sont :
l'interféron standard et l'interféron alpha 2a sous forme
pégylée.
Les interférons ont 3 types d'activité anti
virale :
· Inhibition de la transcription des ARNm et de
l'encapsidation du génome viral ;
· Stimulation des lymphocytes TCD8+ cytotoxiques et
augmentation de l'expression des molécules HLA de classe1 membranaires,
aboutissant à une présentation plus efficace des antigènes
viraux aux lymphocytes T cytotoxiques ;
· Activité anti tumorale.
1.8.1.2.2. Interféron standard
On distingue : l'interféron alpha 2a
(Roféron A®) et l'interféron alpha 2b (Intron A®).
L'interféron entraine une réponse virologique
prolongée (séroconversion stable 24 semaines après
l'arrêt du traitement) dans 20 à 40% des cas [23].
Les posologies recommandées à l'heure actuelle
sont 5 millions d'unités par jour ou 10 millions d'unités trois
fois par semaine, par voie sous cutanée.
La durée du traitement est de 24 semaines dans les cas
d'hépatite chronique B AgHBe positif et au moins 48 semaines en cas
d'AgHBe négatif [23].
Les effets secondaires de l'interféron sont
fréquents et nombreux, mais peu graves et réversibles à
l'arrêt du traitement ; le plus fréquent est le syndrome
grippal, habituellement modéré. D'autres sont plus rares mais
peuvent être graves ; tels que : le syndrome dépressif,
la décompensation d'une psychose préexistante et la dysthyroidie.
Sont également possibles : asthénie, amaigrissement,
alopécie, troubles du sommeil, troubles de la concentration, troubles de
l'humeur, sécheresse cutanée et biologiquement une
neutropénie et une thrombopénie.
1.8.1.2.3. Interféron
pégylé
L'interféron pégylé est
l'interféron standard, conjugué à une molécule de
polyéthylène glycol (PEG). Cette conjugaison permet de diminuer
la clairance rénale de l'IFN, augmentant ainsi la demi-vie plasmatique
de la molécule. La concentration plasmatique est donc plus stable,
permettant une seule injection par semaine.
L'IFN PEG administré en une injection par semaine est
plus efficace, dans le traitement de l'hépatite chronique B AgHBe
positive, que l'IFN standard en trois injections par semaine.
La tolérance est comparable à celle de l'IFN
standard.
Il existe sous le nom d'interféron alpha 2a sous forme
pégylée (Pegasys®) [23].
1.8.1.2.4. Lamivudine
La lamivudine est un analogue nucléosidique qui inhibe
directement l'ADN polymérase du VHB par intermédiaire de son
métabolite triphosphorylé (lamivudine 5'-triphosphate). Elle agit
également par effet terminateur de chaine.
Initialement, elle avait été
développée comme inhibiteur de la transcriptase inverse du VIH,
puis elle s'est révélée efficace à faible
concentration contre le VHB. Elle est commercialisée sous le nom de
Zeffix® dosée à 100 mg.
Les avantages de la lamivudine sont la prise orale par
comprimé à 100 mg, une excellente tolérance, un effet anti
viral rapide.
Son inconvénient majeur est l'apparition de souches
résistantes à la lamivudine par sélection de mutants dans
la région YMDD de la polymérase. Le taux de résistance
dépend de la durée du traitement : 24% à 1 an, 38%
à 2 ans, 50% à 3 ans et 67% à 4 ans, selon LIAW [24].
Une élévation d'un log (facteur 10) de l'ADN
sérique sous traitement par lamivudine doit faire évoquer la
survenue d'une résistance et introduire l'adénofovir en
poursuivant la lamivudine jusqu'à ce que l'adénofovir ait
provoqué une réponse virologique [23].
1.8.1.2.5. Adénofovir
La molécule administrée est l'adénofovir
dipivoxil, commercialisée sous le nom de Hepsera®,
précurseur de l'adénofovir, analogue nucléosidique de
l'adénosine mono phosphate.
« In vivo », l'adénofovir
dipivoxil est métabolisé en adénofovir, lui-même
phosphorylé en adénofovir diphosphate, métabolite actif,
inhibiteur compétitif de l'ADN polymérase qui bloque la
synthèse de l'ADN du VHB [23].
Peu d'études sont actuellement disponibles concernant
l'adénofovir dipivoxil, mais une résistance a été
récemment décrite, par sélection d'un virus mutant au
niveau du domaine D de la polymérase en position 236, par remplacement
d'une asparagine par une thréonine (rtN236T). Ce mutant reste sensible
à la lamivudine.
1.8.1.2.6. Nouveaux antiviraux
De nouvelles molécules sont en cours d'étude
pour le traitement de l'hépatite chronique B : la ténofovir,
l'entécavir, l'emtricitabine, la telbuvidine et la clévudine
[23].
La ténofovir et l'emtricitabine sont déjà
utilisés dans le traitement anti VIH et sont entrain d'être
testés dans le traitement anti VHB.
1.8.1.3. Stratégie
thérapeutique
L'interféron standard est actuellement la
molécule recommandée en première intention dans le
traitement de l'hépatite chronique B [1]. En pratique il est
supplanté par l'INF PEG, étant donné son efficacité
et sa meilleure maniabilité [23].
En cas d'échec ou de contre-indication au traitement
par interféron, la lamivudine ou l'adénofovir doivent être
utilisés.
La durée du traitement par la lamivudine et
l'adénofovir est mal connue. En cas de séroconversion HBe, la
règle est de poursuivre le traitement pendant 3 à 6 mois, afin de
réduire le risque de, réactivation. En l'absence de
séroconversion dans l'hépatite chronique B AgHBe positif ou dans
le cas d'hépatite chronique AgHBe négatif, le traitement doit
être poursuivi tant qu'il est efficace, c'est-à-dire tant qu'il
n'y a pas de réactivation due à une résistance [24].
1.8.1.4. Indications du traitement
antiviral
Le traitement antiviral ne s'envisage que dans le cadre des
hépatites B chroniques. Le principal facteur à prendre en compte
est la gravité de la maladie hépatique, déterminée
par la PBH.
Le traitement est indiqué chez les patients ayant une
activité modérée ou sévère (activité
METAVIR= A2) et/ou une fibrose sévère (fibrose METAVIR=F2).
Les patients ayant une activité hépatique minime
et/une fibrose minime ne doivent pas être traités, mais
surveillés de façon régulière, afin d'instaurer un
traitement en cas d'apparition d'une activité modérée ou
sévère.
Les patients AgHBs positifs avec des manifestations extra
hépatiques doivent être traités si la multiplication est
active et jugée responsable de ces manifestations.
Au stade des complications, les patients doivent être
traités [1, 23].
1.8.2. TRAITEMENT PREVENTIF
Le traitement préventif de l'infection à VHB
repose sur les mesures d'hygiène, l'immunisation passive et la
vaccination.
1.8.2.1. Mesures d'hygiène
Les mesures d'hygiène visent à éviter la
survenue de l'infection à VHB. Les mesures les plus pertinentes
sont : l'utilisation des préservatifs, l'éviction du don de
sang des échantillons positifs pour l'AgHBs, pour les anticorps anti-HBc
ou ayant les transaminases élevés, l'utilisation du
matériel médico chirurgical et dentaire à usage unique ou
correctement stérilisé, le port de gants lors des soins,
programmes de réduction de drogues illicites par voie veineuse et la
proscription absolue du partage interindividuel du matériel pouvant
être en contact avec le sang (brosse à dents, rasoirs,...)
[21].
1.8.2.2. Immunisation passive
L'immunisation passive repose sur l'injection
d'immunoglobulines spécifiques anti-HBs obtenues à partir de
sujets immunisés contre le VHB. Elle confère une protection
immédiate mais transitoire (environ 6 semaines) et permet de
réduire de 75% le risque d'hépatite B chez les patients ayant eu
un contage pour le VBH [21] :
Ø Contamination accidentelle par piqûre ou
blessure par des produits sanguins contenant l'AgHBs dans les 48 heures
suivant ;
Ø Contact sexuel avec un sujet infecté par le
VHB ;
Ø Sujet à risque élevé d'infection
par le VHB (hémodialysés) pour couvrir la période
précédent la protection par la vaccination ;
Ø Transplantation hépatique chez un porteur
chronique de l'AgHBs, en dehors de toute virémie détectable avant
la transplantation ;
Ø Nouveau-né de mère porteuse d'AgHBs.
Les posologies recommandées sont : 500 UI en cas
de contage accidentel, 30 UI/Kg chez le nouveau-né et 10 000 UI
tous les mois chez les greffés hépatiques infectés par le
VHB et pendant une durée prolongée afin de maintenir un taux
d'anticorps supérieur à 500 mUI/ml.
1.8.2.3. Vaccination
La vaccination est pratiquée de façon courante
depuis les années 80 et repose sur l'injection de l'AgHBs
destinée à induire la production d'anticorps anti-HBs
neutralisants. Les vaccins actuellement disponibles sont produits par
génie génétique et contiennent de l'AgHBs recombinant et
éventuellement d'autres sous unités de l'enveloppe virale.
Les vaccins actuellement disponibles en France sont
[23] :
Ø GENHEVAC B®constitué d'une suspension
inactivée et purifiée de l'AgHBs contenant les protéines S
et pré-s ;
Ø ENGERIX B contenant l'AgHBs purifié ;
Ø TWINRIX est un vaccin combiné, contre
l'hépatite B et l'hépatite A ;
Ø INFANRIX HEXA, est un vaccin combiné
hexavalent contre la diphtérie, le tétanos, la coqueluche, la
poliomyélite et l'haemophilus influenzae b.
La vaccination est indiquée chez tous les
nouveau-nés, tous les sujets à risque d'infection par le
VHB (toxicomanes, sujet avec multiples partenaires sexuels, acteurs des
soins médicaux) et les femmes enceintes des pays où la
vaccination anti-VHB n'est pas pratiquée [23].
Les vaccins sont administrés par voie intramusculaire
selon un schéma classique de 3 doses (0, 1 et 6 mois). Au-delà de
ces 3 injections, il n'est plus nécessaire d'effectuer des rappels
systématiques, la diminution du titre des anticorps anti-HBs sous le
seuil de 10 mUI/ml ne signant pas l'absence de protection.
1.9. GENOTYPES DU VHB
1.9.1. CONCEPTS GENERAUX
Historiquement, la variabilité du VHB a
été évaluée par des techniques sérologiques
utilisant des anticorps monoclonaux dirigés contre l'AgHBs, aboutissant
à une classification sérologique. Celle-ci est définie par
un déterminant antigénique « a »
commun aux différents sous types et deux paires de déterminant
exclusifs d/y, w/r. Après des divisions en 4 types majeurs
(adw, adr, ayw et ayr), un total de 9 sous-types a
été identifié (adw1, adw2, ayw1, ayw2, ayw3,
ayw4, adwyq+ et ayr), en fonction des
différents déterminants liés à des mutations
nucléotidiques d'une région immunologiquement compétente
de l'AgHBs [26].
Grâce aux techniques de séquençage complet
de l'ADN viral, un système de classification génétique du
VHB a été établi. On dénomme par VHB un groupe
complexe de virus relativement proches ayant une spécificité
d'hôte et caractérisés par la présence d'au moins un
des critères suivants :
· Une divergence intergroupe de 8% ou plus de la
séquence nucléotidique dans tout le génome ;
· Une divergence de 4,1% ou plus dans le gène de
surface (pré-S1, pré-S2 et S) [26].
A l'heure actuelle, on définit chez l'homme 8
génotypes du VHB, dénommés A, B, C, D, E, F, G et H. bien
qu'il existe de nombreuses corrélations entre sous-types et
génotypes. C'est le génotype qui est maintenant utilisé
pour étudier la variabilité génétique.
1.9.2. MECANISMES DE VARIATION GENETIQUE
La variabilité génétique du VHB peut
être expliquée par la résultante d'un équilibre
entre plusieurs facteurs [26] :
Ø Les erreurs d'incorporation de la polymérase
non corrigées par l'absence d'activité 3'-5' exonucléase,
le nombre total d'erreurs étant de 1010 paires de bases par
jour ;
Ø L'espace de la réplication virale,
correspondant à la capacité d'intégration par les
hépatocytes de nouveaux ADN superenroulés ;
Ø La pression de sélection exercée lors
de certains traitements ;
Ø Le fitness, c'est-à-dire le pouvoir infectieux
propre à chaque souche virale.
La majorité des particules virales ainsi formées
sont défectives et ne peuvent pas se répliquer. Certains variants
viraux sont, par contre, capables d'infecter de nouvelles cellules, de se
multiplier et d'être sélectionnés.
L'accumulation des mutations, la sélection des
séquences virales les mieux adaptées, la transmission des virus
correspondants au sein d'aires géographiques ou de groupes
épidémiologiques déterminés ont conduit à la
divergence progressive à partir de leur ancêtre viral commun selon
un processus darwinien classique [26].
1.9.3. METHODES DE GENOTYPAGE
Il existe actuellement 4 méthodes principales pour
déterminer les génotypes du VHB. Les deux plus souvent
utilisées sont l'étude du polymorphisme de restriction (RFLP pour
Restriction Fragment Length Polymorphisms) et le LiPA (Line Probe
Assay).
1.9.3.1. Séquençage direct
L'ADN viral est extrait du sérum du patient, puis
amplifié par une méthode de PCR utilisant des amorces dans des
régions spécifiques. Le séquençage est
réalisé sur les produits de la PCR et les séquences sont
comparées aux séquences correspondant connues pour chaque
génotype. C'est une méthode très sensible, actuellement
considérée comme référence en termes de
génotypage du VHB.
Ses limites sont un coût élevé et la
nécessité d'un travail intense, limitant son utilisation à
grande échelle en clinique [26].
1.9.3.2. Etude du polymorphisme de restriction
Comme lors du séquençage direct, l'ADN viral est
extrait du sérum et amplifié en utilisant une méthode de
PCR. Les produits de la PCR, contenant des régions spécifiques du
génotype viral sont ensuite digérés par des enzymes de
restriction et migrent par électrophorèse sur gel d'agarose.
Après coloration, les profils de migration sont comparés aux
profils de migration connus spécifiques de chaque génotype.
L'étude du polymorphisme de restriction est plus simple
à mettre en évidence que le séquençage direct. Elle
est donc plus largement utilisée, notamment lors des études
épidémiologiques [26].
1.9.3.3. Méthode ELISA (Enzyme-Linked
Immunosorbent Assay)
C'est une méthode immunologique qui utilise un
anticorps fixe, dirigé contre le déterminant commun a de l'AgHBs,
afin de capturer l'AgHBs dans le prélèvement. On ajoute ensuite
un anticorps monoclonal spécifique du génotype recherché,
afin de typer le génotype viral contenu dans l'échantillon.
Il existe une étroite corrélation entre le
niveau de détection de l'AgHBs et la capacité de
déterminer le génotype par ELISA. Bien que les kits ELISA soient
simples d'utilisation et peu couteux, il est possible que les patients avec de
faibles taux d'AgHBs circulant ne puissent bénéficier de cette
méthode pour déterminer le génotype de leur virus [26].
1.9.3.4. Méthode LiPA
C'est une technique d'hybridation après amplification
par PCR. Des séquences spécifiques des génotypes sont
connues dans les régions du gène de l'AgHBs. Des sondes
complémentaires de ces régions spécifiques sont
synthétisées et fixées sur un support. Les produits de la
PCR du gène S sont mis en présence des bandelettes
réactives et vont s'hybrider en fonction de leur
complémentarité avec les sondes. On peut ainsi déterminer
les génotypes viraux en comparant les profils d'hybridation avec des
abaques références. C'est une technique fiable et disponible
[26].
1.9.4. DONNEES EPIDEMIOLOGIQUES
Les génotypes du VHB sont retrouvés de
façon ubiquitaire dans le monde, mais il est maintenant établit
que leur distribution varie en fonction des données
géographiques. Ainsi, on peut établir une cartographie (Figure 5)
reflétant la distribution des génotypes prédominants dans
chaque région du globe [27].

Figure 6. Répartition
géographique des principaux génotypes du VHB [27]
1.9.5. DIFFERENCES VIROLOGIQUES
Différentes études se sont
intéressées aux mutations du VHB et à leur
éventuelle corrélation avec le génotype viral. Ce sont les
mutations pré-core et BCP responsables de l'hépatite chronique
AgHBe positif, qui ont fait l'objet du plus grand nombre d'études.
1.9.5.1. Génotypes et mutation
pré-core
Dans la séquence d'ADN du VHB, la guanosine en position
1896 s'apparie avec le nucléotide en position 1858 (codon 15) pour
former une structure circulaire hautement conservée, signal
d'encapsidation du virus.
Chez les souches sauvages du VHB de génotypes B, C, D,
E et G, ce nucléotide en position 1858 est une thymidine qui forme, avec
la guanosine en position 1896 de la séquence d'encapsidation, une
structure circulaire peu stable. La mutation (G A) en position 1896
crée un codon stop responsable de l'arrêt de l'expression de
l'AgHBe, mais stabilise la structure circulaire de l'encapsidation du virus par
appariement (T-A), ce qui favorise la multiplication virale.
Inversement chez les souches sauvages de VHB de
génotypes A, F, H et quelques C, le nucléotide en position 1858
est une cytidine qui s'apparie de façon stable avec la guanosine en
position 1896 (C-G). La mutation (G A) en position 1896
déstabilise la structure et, est rarement observée chez les
patients infectés par les génotypes A, F et H, à moins
qu'elle ne survienne de façon concomitante avec une autre mutation comme
C1858T [28].
Chu et al ont montré que sur les 694 patients
de leur étude aux Etats Unis en 2003, 27% présentaient des
mutations pré-core. Ces mutations étaient plus fréquentes
chez les patients porteurs de virus de génotypes D (73%) et B (46%) et
rares chez les porteurs de virus de génotype A (3%) [29].
1.9.5.2. Génotypes et mutation
BCP
Les mutations BCP présentent une double substitution
nucléotidique A1762T et G1764A. Elles ont une sécrétion de
l'AgHBe réduite de 70%. Ces mutations n'affectent pas la transcription
des ARN prégénomiques ni la traduction des protéines de
core ou de la polymérase. Au contraire, elles contribuent à
accroitre la réplication virale, d'une part, en levant l'effet
inhibiteur de l'AgHBe sur la réplication virale, d'autre part, en
supprimant la synthèse des ARN messagers pré-core et core, au
profit de la synthèse des ARN prégénomiques [4].
La coexistence des mutations pré-core et BCP est
possible chez un même individu, notamment en cas d'infection par un virus
de génotype D [29].
1.9.6. CONSEQUENCES CLINIQUES
Actuellement, peu d'études sont informatives sur les
conséquences cliniques du génotypage du VHB. Des analyses
comparant un grand nombre de patients infectés par les principaux
génotypes et soumis aux mêmes protocoles thérapeutiques et
de surveillance, sont nécessaires pour prouver l'utilité du
génotypage du VHB en pratique clinique. Fréquemment, les
études asiatiques comparent les génotypes B et C, alors que les
études occidentales comparent le plus souvent les génotypes A et
D.
Il est clairement établi que tous les génotypes
viraux sont potentiellement infectants pour les individus et peuvent conduire
à une hépatite aiguë ou chronique, à la cirrhose, au
CHC et à la mort. Le taux de progression de la maladie et l'importance
des lésions hépatiques pourraient varier en fonction des
génotypes viraux, mais aussi être influencés par des
facteurs environnementaux et par l'hôte [28].
Wai et al ont démontré que l'infection
par un virus de génotype C est associée à une maladie
hépatique plus sévère que celle observée au cours
de l'infection par un virus de génotype B. Ainsi, dans le cas d'un
génotype C, les taux de transaminases et de l'ADN viral sont plus
élevés. De même, l'histologie hépatique est plus
sévère et le risque de cirrhose et plus élevé [26].
Par ailleurs, une étude réalisée à Hong Kong
révèle que le taux de séroconversion HBe est plus
important dans le cas d'un génotype B que C [30].
Sanchez-Tapias et al ont montré que
l'infection chronique due au virus au génotype A aurait un meilleur
pronostic que celles dues aux génotypes D et F. Les réponses
biochimiques et virologiques se rencontrent plus fréquemment chez les
sujets infectés par un virus au génotype A que chez ceux
infectés par le virus au génotype D ou F [31].
En France, une étude récente concernant 308
patients co-infectés par le VIH révèle que le fait
d'être infecté par un virus au génotype G est un facteur
d'évolution rapide vers la fibrose hépatique [32].
1.9.7. GENOTYPES ET TRAITEMENT
1.9.7.1. Génotypes et traitement par
l'interféron
Il existe des différences dans la réponse
à l'INF en fonction du génotype viral. Les données
disponibles comparent les génotypes A et D en rapport avec la
distribution géographique.
En effet, le taux de réponse au traitement par l'INF
est meilleur chez les patients infectés par un virus au génotype
A que ceux infectés par un virus au génotype D. De même, la
réponse au traitement par l'INF est meilleure chez les patients
infectés par un virus au génotype B que ceux infectés par
un virus au génotype C [33].
1.9.7.2. Génotypes et traitement par
lamivudine
A ce jour, peu d'études comparent l'efficacité
de la lamivudine en fonction du génotype. Ces études ne sont pas
comparables compte tenu du faible nombre de patients et des différents
protocoles thérapeutiques adoptés et les résultats sont
contradictoires [34].
Il est donc impossible d'affirmer qu'il existe des
différences dans la réponse au traitement de l'hépatite
chronique B par la lamivudine en fonction du génotype viral.
La résistance à la lamivudine en fonction du
génotype viral est également restreinte à quelques
études qui ne permettent pas de conclure à un rôle du
génotype à la survenue de mutants YMDD [34].
1.9.7.3. Génotypes et traitement par
adénofovir
La réponse virologique au traitement par
adénofovir ne semble pas être influencée par le
génotype du virus responsable de l'hépatite chronique B, comme en
témoignent les résultats de Westland et coll. Concernant 694
patients traités par 10 mg d'adénofovir pendant 48 semaines
[35].
1. CADRE D'ETUDE
Les données épidémiologiques ont
été collectées d'une part par l'intermédiaire d'une
fiche d'enquête standardisée (annexe1), élaborée
spécifiquement pour notre étude; d'autre part, par
l'intermédiaire des registres au Centre inter départemental de
Transfusion Sanguine (CIDTS) de Pointe-Noire.
PATIENTS ET METHODES
Les prélèvements ont été
analysés d'une part au laboratoire de l'Hôpital
Général de Loandjili (HGL) et d'autre part au laboratoire de
virologie/UPRES EA 3610, Faculté de Médecine, Université
Lille II, CHRU Lille, Centre de Biologie Pathologie et Parc EURASANTE.
1.1. Laboratoire de l'HGL
L'Hôpital Général de Loandjili (HGL) est
situé dans l'arrondissement 4 de Pointe-Noire. Il dispose de quatorze
(14) services cliniques et de trois (3) services médico-techniques,
parmi lesquels le laboratoire, où notre étude a été
réalisée.
Le Laboratoire comporte 7 unités (anatomo-pathologie,
virologie, hématologie, biochimie, parasitologie-mycologie,
séro-immunologie et l'unité de bactériologie). Il est
dirigé par le Docteur Donatien MOUKASSA, anatomo-pathologiste.
| 


