CHAPITRE 2
RESOLUTION DE L'EQUATION DE
SCHRÖDINGER NUCLEAIRE.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
2.1. INTRODUCTION.
Après avoir déterminé les puits de
potentiel sur lesquels se meuvent les
électrons, il est tout aussi très
intéressant de maîtriser les mouvements des noyaux. Dans cette
section, la théorie des groupes nous sera d'une très grande
utilité.
2.2. RESOLUTION DE L'EQUATION DE SCHRÖDINGER
NUCLEAIRE PAR
LA METHODE DES PERTUBATIONS.
L'étude de la dynamique des noyaux revient à la
résolution l'équation de Schrödinger nucléaire (1.21)
:
? T N + V j ( ) ? Ø Nj
( ) = E T Ø Nj ( ) .
R R R
? ?
Pour résoudre cette équation, on fait en plus
de l'approximation de Born-Oppenheimer, une deuxième approximation
d'ordre zéro qui suppose que le mouvement de rotation de la
molécule est indépendant de ses mouvements de vibration ; de
sorte que l'on peut étudier la vibration de la molécule en ne
tenant pas compte de sa rotation, puis la rotation globale de la
molécule supposée indéformable.
2.2.1. Vibrations moléculaires.
2.2.1.1. Cas simple d'une molécule
diatomique.
2 2
? ?
h ?
? - 2 U(R) n E
+ ? Ø = n
Ø . (2.1)
? ì ?
2 R ?
Considérons le cas d'une molécule diatomique
qu'on assimilera à un système constitué de deux atomes de
masses ponctuelles M1 et M2 séparées par une distance R et
interagissant par l'intermédiaire d'un potentiel U(R). Le système
ainsi formé a une énergie totale E et vérifie
l'équation de Schrödinger suivante :
Le premier terme dans (2.1) est l'énergie
cinétique de vibration ; est la masse réduite
ì
du système.
Un développement en série de Taylor de U au
voisinage de son minimum ( R = R e)
donne :
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
(2.2)
? ?
dU 1 d U
? ?
2 2 1 d U
? ?
3
3
U(R R )
- = ? ? - + ? ? -
( )
R R ( )
R R + ? ? -
( )
R R + ...
e e 2 e 3 e
? ?
dR 2! dR
R ? ? 3! dR
? ?
e R R

e e
2! ?dR
1 d 2 U (R -R e)2 + 1 d3U1 ( R
-R e)3 +...
2 R 3! ? dR3 R
e e
Les mouvements moléculaires étant de faible
amplitude, on peut, dans l'approximation harmonique, négliger les termes
de degré supérieur à deux. Ainsi, on a :
U(R - R e ) = 21k ( R
-Re)2
Où k = C1.2U
dR2 est la constante de force harmonique. Ainsi, la
résolution de

Re
l'équation (2.1) conduit aux solutions suivantes :
[ Ø n p) C p e 2 á ( R -Re) 2H p (
á ( R - Re ))
(2.4)
E ( p) = ( p+ 12 )hù
1
Lì 1 dRd 2 U2 ?2
03 = Avec p ? 0 , =pulsation de l'oscillateur. ; H
p=polynôme
Re
d'Hermite ;
( 2 p p! á ð) 2
ìù
= Constante de normalisation et .
á =
h
Cp =

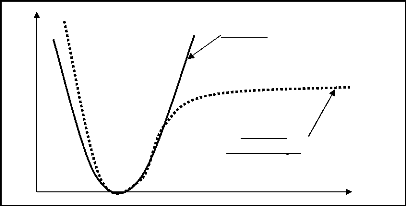
Potentiel
Potentiel
anharmonique
Figure 2.1 : Potentiel harmonique et
anharmonique.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
Physiquement, le potentiel harmonique ne parvient pas
à expliquer la dissociation des molécules et certaines
transitions vibroniques. En effet, l'approximation harmonique devient
très vite insuffisante dès que l'on s'éloigne de la
position d'équilibre. D'où l'idée de l'approximation
anharmonique.
2.2.1.2. Potentiel anharmonique.
Pour un tel potentiel, on ne néglige plus les termes
de degré supérieur à 2 dans l'équation (2.2). Toute
fois, comme ils sont très inférieures au terme quadratique, on
peut considérer la correction qu'ils apportent à l'hamiltonien
total du système comme une perturbation stationnaire. Ainsi, en
première approximation, on peut écrire :
1 2 1 d U
? ?
3
3
U(R R ) k R R
- ( )
- + ? ? -
( )
R R
e e 3 e
2 6 dR
? ? R e
Par suite, au second ordre de perturbation, on a :
|
. (2.5)
|
|
( )
p ? ? ?
1 1
E = ? + ? ù - ? +
p h p
? ? ?
2 2
? ? ù x
h . (2.6)
e
?
2
Où xe est une constante positive
appelée constante d'anharmonicité. L'écart entre deux
niveaux consécutifs d'énergie n'est plus constant et vaut :
E E
( p ) ( p 1 )
- ? ?
- = ù ? - ? +
h 1 x p
e
? ?
Ainsi, les niveaux se resserrent lorsqu'on monte en
énergie.
Une des formes analytiques de potentiel anharmoniques la plus
couramment utilisée est le potentiel de Morse[21] (fig. 2.2)
:
U R D e 1 e-â -
( e ) 2
R R
( ) = ? - ? ? . (2.8)
?
.
hùe
=
x e
(2.11)
4D
e
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
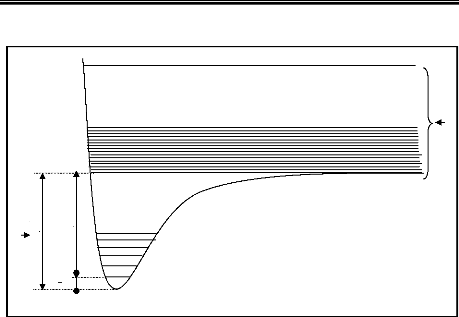
.
.
(a)
.
.
.
De
D0
.
(b)
2 hù
1
Figure 2.2 : Potentiel de Morse. (a)
Continuum, (b) Limite de dissociation
Où De est l'énergie de dissociation et
â une constante caractéristique de la molécule
considérée.
En faisant un développement limité à
l'ordre 3 au voisinage de la position d'équilibre Re, on
trouve :
( ) ( ) ( ) ( ( ) )
â 3
2 2 3 3
U R D e R R e
= â - - R R e
- + Ï -
R R . (2.9)
e
2
En identifiant cette relation à la relation (2.5), on
arrive à :
1
? ì ? 2
â = ? ? ù . (2.10)
e
? ?
2De
Et l'énergie du système est donnée par la
relation (2.6) avec
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
2.2.1.3. Vibrations moléculaires en
mécanique quantique[22].
Dans une molécule polyatomique, les atomes
constitutifs subissent des oscillations autour de leur position
d'équilibre. Au cours de ces oscillations, les longueurs des liaisons et
les angles intramoléculaires changent de façon
périodique.
En posant

q i = Miri,
n
M M
% = +? m
i i i
. (2.12)
i 1
=
L'énergie totale de vibration s'écrit :
N 1 N
= ? % r & = ? & qi .
(2.13)
1 2 2
E M
V i i
2 = 2 =
i 1 i 1
L'énergie potentiel de vibration V(q), où q
désigne l'ensemble des coordonnées qi, peut
être
développée en série de Taylor au voisinage de la position
d'équilibre de chaque
qi0
atome ; soit :
N ? ? ?
V 1 N N ? ? ?
2 V
V q V q
( ) ( )
= + ? ? +
? q ?? ? ? +
q q ...
0 i i j
i 1
= q
? ? ? 2
i i 1 j 1 ? ? ? ? ? ?
q q
= =
q i j
i0 q i0
|
(2.14)
|
|
En prenant l'origine de l'énergie potentiel à
l'équilibre, on a ; la courbe
V ( q 0 ) = 0
d'énergie potentiel passe par un minimum
pourqi = q 0 . Ainsi, le second terme du
i
membre de droite de (2.14) est nul. En se limitant à
l'approximation harmonique, on a :
N N ? ? ?
2
1 V
V q
( ) = ?? á q q , á = ? ?
ij i j ij
2 i 1 j 1
= = ? ? ? ? ? ?
q q
i j q
i0
|
. (2.15)
|
|
Pour étudier complètement le mouvement
vibratoire d'un système, nous devons préciser ses
coordonnées normales, ses coordonnées internes et ses
coordonnées de symétrie.
2.2.1.3.1 Coordonnées normales
Les coordonnées normales sont les nouvelles
coordonnées qui conservent la forme
Qi
de l'énergie cinétique, tout en permettant
d'exprimer l'énergie potentiel sous une forme quadratique. En effet,
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
1 1
2 2
E = ? ?
q & = Q&
V i i
2 2
i i
1 1 2 2
V q
( ) = ? á q q = ? ù Q
ij i j k k
2 2
i, j k
(2.16)
?
Où est une fréquence propre du système.
Dans ce cas, une énergie propre du
ùk
système est donnée par
3N 6
-? ?
1
E = ? + ? ù +
? h ? ? xij
n
k k
k 1 ? ?
2
= j j i
<
L'intérêt des coordonnées normales
réside dans le fait qu'elles permettent de réaliser une
représentation irréductible du groupe de symétrie du
système étudié. En effet, à chaque fréquence
propre du système, on peut toujours associer une coordonnée
normale. Ce qui caractérise un mode propre de vibration précis,
qu'on peut aisément associer à une représentation
irréductible du groupe de symétrie du système global. On
comprend donc que les coordonnées normales d'un système
permettent de classifier ses vibrations. Mais, la détermination
explicite de ces coordonnées normales est assez épineuse. Pour
s'affranchir de cette difficulté, on utilisera la théorie des
groupes.
2.2.1.3.2. Classification des vibrations
moléculaires.
Pour classifier les vibrations moléculaires, on
procède comme suit :
· On détermine la représentation totale
engendrée par les 3N coordonnées de déplacement des atomes
de la molécule.
· On détermine la représentation vibratoire
totale v en éliminant dans les
représentations associées aux translations et
rotations d'ensemble.
· On décompose ensuite v en
représentations irréductibles.
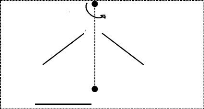
óv
C2
O
H H
Figure 2.3 La molécule d'eau
Etudions par exemple le cas de la molécule d'eau (H2O)
dont la structure géométrique est représentée par
la figure 2.3.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
Compte tenu de ses éléments de symétrie,
cette molécule appartient au groupe C2v. Sa
représentation vibratoire totale est donc :
Ainsi, T = 3A 1 ? A 2 ? 2B 1 ? 3B2 (2.17)
La représentation engendrée par
les
translations et les rotations d'ensemble
C 2v
E
|
C 2
|
óv
|
'
ó v
|
T
|
9
|
-1
|
3
|
-1
|
|
est :
T + R = A 1 ? A 2 ? 2B 1 ? 2B2 (2.18)
Par suite, la représentation vibratoire totale est : V =
2A1 ? B2 (2.19)
On conclut donc que la molécule d'eau a :
9 2 modes de vibrations complètement symétriques
(A1).
9 1 mode de vibration complètement antisymétrique
(B1).
Parlant des modes de vibrations, on distingue :
9 Stretching mode (allongement)
9 Bending mode (courbure)
9 Twisting mode (torsion)
9 Wagging mode (hochement)
9 Scissoring mode (cisaillement)
9 Rocking mode (rotation plane)
De part cet exemple, on constate que la détermination
du nombre de fréquences fondamentales d'une molécule ainsi que
leur classement selon le type de symétrie correspondant aux
représentations irréductibles contenues dans la
représentation vibratoire totale, ne nous apprend rien sur le mouvement
réel des atomes. Il faut donc penser à introduire les
coordonnées internes et les coordonnées de symétrie.
2.2.1.3.3. Coordonnées internes.
Les déplacements des atomes dans une molécule
peuvent en effet être repérés en
considérant les
allongements des liaisons, ainsi que les variations des angles entre
qi â i
les axes de liaison des atomes. Ces variations de longueur et
d'angle sont appelées coordonnées internes de la
molécule.
Pour illustration, reprenons l'exemple du H2O.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
H H
Figure 2.4. coordonnées internes de la
molécule d'eau.
O
â
q1 q2
Ainsi, on a : v = q ? â , (2.20)
avec q = A 1 + B 2 et â =
A1. (2.21)
2.2.1.3.4. Coordonnées de
symétrie.
Les coordonnées de symétrie sont des fonctions
de base des représentations figurant dans une décomposition
canonique. Elles sont des combinaisons linéaires d'un seul type de
coordonnées internes. Elles ont l'avantage de livrer le secret des
mouvements réel des atomes dans une symétrie donnée.
Pour déterminer ces coordonnées de
symétrie, on applique sur tous les coordonnées internes,
l'opérateur projecteur Pá défini par :
n
á = ? ÷
á
P
g G
|
*
( ) ( )
á G
|
.
|
(2.22)
|
|
Où ÷ est le caractère associé à
la représentation irréductible du groupe de
( á )
á
symétrie G ; ( G) est le caractère de la
représentation totale de ce groupe ; ná est la
dimension de á et g est l'ordre du groupe G.
Ainsi, les coordonnées de symétrie de la
molécule de H2O sont :
( )
q q
1 + 2
2
? ? S A
2 ( ) = â
1
1
( ) ( )
= q q
-
1 2
2
1
S A
1 ( ) =
1
?
?
?
?
S A
3 1
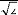
(2.23)
Maintenant, on sait comment les atomes bougent dans un mode de
vibration donnée.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
2.2.2. Rotations moléculaires (rotateur
rigide)
2.2.2.1. Molécules linéaires.
Si on assimile la molécule à un rotateur rigide,
l'Hamiltonien Hr relatif à la rotation s'écrit :
I est le moment d'inertie du système par rapport à
son centre d'inertie G. L étant
l'opérateur moment cinétique dont les composantes
, , dans un repère mobile
L x L y L z
(Gxyz) et , , dans un repère fixe (OXYZ) vérifient
la relation :
LX LY LZ
L L L L L L L
2 = + + = + + Z
2 2 2 2 2 2 . (2.25)
x y z X Y
{ }
L , L z
2 Constitue un E.C.O.C. Donc les fonctions J,M , sont
les bonnes fonctions

propres associées à Hr ; et on a :
H r J,M = E r J,M . (2.26)
En remplaçant Hr par son expression en (2.24),
on obtient :
E J = hcB eJ(J + 1). (2.27)
h
Où =
B est la constante de rotation.
e 8 cI
ð 2
Pour une valeur de J, il existe (2J+1) harmoniques
sphériques è ?
Y , =-
M ( ) . Donc
J M J,...,J
chaque valeur propre EJ est (2J+1) fois
dégénérée.
2.2.2.2. Molécules non
linéaires.
Si on assimile la molécule à un rotateur rigide,
l'Hamiltonien Hr relatif à la rotation s'écrit :
H r
2 2 2
1? L L L ? x y

= + + z . (2.28)
2 I I I
? ?
? xx yy zz ?
Par ailleurs, { }
L , L z , L
Z
2 est un E.C.O.C. D'où la base J,K,M est une
base propre
associée à Hr telle que :
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.

J 0,1,...
=
2
? J,K,M J J 1 J,K,M
2
L = +
( ) h


? L J,K,M K J,K,M
?
?
??
(2.29)
K J,...J
=-
= h
z


M J,...J
=-
LJ,K,M M J,K,M
= h
Z
LZ
Les fonctions propres correspondant aux vecteurs propres
simultanés de , et
L 2 Lz
sont de la forme [23] :
Ö ( ) È è
J iK
J,K,M , ,
è ? ÷ = N J,K,M K,M e ÷ eiM ?
( )
|
.
|
(2.30)
|
|
Où est la constante de normation, et est un
polynôme de Legendre. ,
J
N J,K,M È K , M Ixx
Iyy
|
et sont les moments principaux d'inertie dans les directions
(Gx), (Gy) et (Gz)
Izz
|
|
respectivement. Ils permettent de caractériser la
molécule :
· Si les trois moments principaux d'inertie sont
égaux, la molécule est une toupie sphérique.
· Si I xx = I yy ? I zz , la molécule est
une toupie symétrique.
· Si I xx ? I yy ? I zz, la molécule est
dite toupie asymétrique.
Par ailleurs, l'énergie de rotation du système est
donnée par l'équation :


H r J,K,M = E J J,K,M . (2.31)
Ainsi,
V' Dans le cas d'une toupie
sphérique, on a :
E J = hcB e J ( J + 1). (2.32)


Et, pour chaque valeur de J ; K et M prennent chacun (2J+1)
valeurs. Par suite, à
chaque niveau d'énergie EJ correspond
(2J+1)2 valeurs de J,K,M différentes. EJ
est alors (2J+1)2 fois
dégénérée.
V' Dans le cas d'une toupie
symétrique, on a :
E J, K hc ? B e J J 1 A e B e
K

= ( ) ( ) 2
+ + - ? . (2.33)
? ?
B
|
|
h
|
|
h
et sont les constantes rotationnelles à
l'équilibre.
A =
e 8 cI
ð 2
zz
|
|
|
|
|
xx
|
|

Pour une valeur donnée de J, le nombre quantique M peut
prendre (2J+1)
valeurs différentes ; et comme EJ, K dépend de
la valeur absolue de K, il lui
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.


correspond 2(2J+1) valeurs de J,K,M différentes. E J, K
est donc 2(2J+1)
fois dégénérée.
v' Dans le cas d'une toupie
asymétrique, plusieurs calculs ont été faits
pour obtenir une formule donnant les énergies EJ. Nous donnons
l'expression proposée par Wang[24] :
? 1 1
E hc B C J J 1 A
J ( e
= ? + e ) ( ) e ( e
? ? ?
+ + - B C W
+ e ) ? ? , (2.34)
? ô
? 2 ? 2 ? ?
h
avec C = .
yy
e 8 cI
ð 2
Pour J donnée, prend (2J+1) valeurs différentes ;
et par conséquent, on a (2J+1)
Wô
états différents [23].
2.2.3. Solution du problème
nucléaire.
Le mouvement nucléaire peut se décomposer en
vibrations, rotations et translations. Dans un premier temps, on se place dans
le référentiel du centre de masse de la molécule. Ce qui
revient à ne plus percevoir le mouvement de translation. Par ailleurs,
les vibrations et les rotations sont faiblement couplées. On
néglige donc ce couplage. Ce qui revient à exprimer la fonction
d'onde nucléaire comme un produit de fonctions d'onde vibrationnelle et
rotationnelle.
La fonction d'onde vibrationnelle est le produit d'une vibration
d'élongation et d'une vibration angulaire.
Ainsi, la fonction d'onde vibrationnelle peut s'exprimer :
2
( ( ) ) ( ) ( )
1 R R
á - e l
Ö = 2
vib A H n á -
R R e e P n cos ã . (2.35)
Par suite, la fonction d'onde nucléaire pourra être
écrite comme suit :
2
( ( ) ) ( ) ( ) ( )
2 1 R R
á - e l J iK
N A H n R R e e ã È è
P n cos K,M e e
÷ iM ?
Ö = á - . (2.36)
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
| 

