
Projet de Fin d'Etudes
Présentée à :
L'Université Chouaïb Doukkali
Faculté Des Sciences
Licence Fondamentale en Matière de
Chimie
Spécialité: Chimie Physique
Les méthodes QSAR/QSPR et identification
de
nouveaux médicaments: SARS_CoV-2
Par :
Assia REGRAGUI et Ibtissam BENCHIHEB
Sous la direction de :
Pr. Mohammed Salah
Date de soutenance : 25 septembre 2020 à la
Faculté Des Sciences d'El Jadida, Université
Chouaib Doukkali
Résumé
Depuis l'antiquité le monde a connu une apparition
fréquente de maladies et épidémies par des virus. Plus
récemment, le monde a connu une propagation sévère et
rapide de l'épidémie causé par le virus SARS-CoV-2 depuis
la fin de l'année 2019 jusqu'à maintenant. Cette infection virale
a touché et tué un grand nombre de personnes dans monde entier.
Tout cela rendre la découverte de nouveaux médicaments pour
lutter contre ces maladies et épidémies munie d'une grande
importance. Ce présent rapport a un objectif principal qui consiste dans
une part à faire une introduction sur la modélisation avec les
méthodes QSAR/QSPR et d'une autre part de faire une identification
d'approches visant la découverte de nouveaux médicaments,
différentes types de médecines et les médicaments qui
peuvent prévenir et lutter contre le virus SARS-CoV-2.
Remerciements
Au terme de ce projet de fin d'étude, nous tenons à
exprimer nos respects et nos sincère remerciement à
Monsieur Mohammed Salah pour nous 'avoir encadré et
fait de leurs mieux afin de nous aider.
Nous chers parents, que nul remerciement ne puisse exprimer nos
sincères sentiments, Pour leur patience illimitée, leur
encouragement contenu, leur aide, en témoignage de notre profond amour
et respect pour leurs grands sacrifices.
Nos chers grands parents, pour me assistant.
Nos chers frères pour leur grand amour et leur soutien
qu'ils trouvent ici l'expression de notre haute gratitude.
Et à toutes nos familles et à tous ceux que nous
aimons.
Assia REGRAGUI et Ibtissam BENCHIHEB
Table des matières
Introduction générale 1
CHAPITRE 1 : Modélisation QSPR/QSAR 2
1. Les méthodes QSAR/QSPR 2
2. Méthodologie générale d'une
étude QSAR/QSPR 4
2.1 Les descripteurs moléculaires 4
2.1.1 Définition 4
2.1.2 Types de descripteurs 5
a. Les descripteurs 0D 5
b. Les descripteurs 1D 5
c. Les descripteurs 2D 6
d. Les descripteurs 3D 7
e. Les descripteurs locaux des propriétés
surfaciques moléculaires 8
f. Les descripteurs quantiques/électroniques 8
2.2 Méthodes d'analyse de données 8
2.2.1 Méthodes basées sur les descripteurs 9
a. Approches linéaires 9
b. La régression linéaire multiple (MLR) 9
c. La méthode de régression des moindres
carrés partiels 10
d. Approches non linéaires 10
e. Réseaux de neurones artificiels 10
f. Arbre de décision 11
2.3 Interprétation et validation d'un modèle
QSPR/QSAR 12
2.3.1 Validation interne 12
2.3.2 Validation externe 12
3. Applications 13
4. Conclusion 13
CHAPITRE 2 : Conception et développement de
nouveaux médicaments Contre le virus
" SARS-CoV-2" 14
1. Introduction 14
2. Les approches adoptées dans la recherche de
nouveaux médicaments de "SARS-
CoV-2". 15
2.1 Approche basée sur la médecine
traditionnelle 15
2.2 Approche basée sur les fragments 15
2.3 Approche basée sur la carte de connectivité
(Connectivity Map) 16
2.4 Approche basée sur la médecine in silico
16
3. Identification de médicaments antibiotiques de
"SARS-CoV-2" en l'absence d'une
thérapeutique antivirale et d'un vaccin
spécifiques 16
3.1 Des médicaments antibiotiques 16
3.1.1 Amoxicilline 16
3.1.2 Azithromycine 17
3.1.3 Fluoroquinolones 18
3.2 Des médicaments antiviraux 18
3.2.1 Lopinavir / Ritonavir 18
3.2.2 Ribavirin 19
3.2.3 Favipiravid (T-705) 19
3.2.4 Remdesivir 20
3.2.5 Oseltamivir 21
3.2.6 Chloroquine 21
3.2.7 Interférons 22
3.3 Les corticostéroïdes (méthylprednisolone)
22
4. Identification de nouveaux médicaments herbal
de "SARS-CoV-2" 23
4.1 Les médicaments anti-coronavirus à base de la
médecine traditionnelle Chenoise 23
4.1.1 Radix astragali (Huangqi) 24
4.1.2 Glycyrrhizae Radix Et Rhizoma (Gancao) 25
4.1.3 Radix saposhnikoviae (Fangfeng) 25
4.1.4 Rhizoma Atractylodis Macrocephalae (Baizhu) 26
4.1.5 Forsythiae Fructus (Lianqiao) 26
4.1.6 Lonicerae japonicae flos (Jinyinhua) 27
4.2 Les médicaments anti-coronavirus à base de la
médecine traditionnelle Marocaine 27
4.2.1 Digitoxigenin 28
4.2.2 â-Eudesmol 29
4.2.3 Crocin 29
5. Conclusion 31
Conclusion générale 32
6. Bibliographie 33
Liste des figures
Figure 1. Schéma d'élaboration
et validation d'un modèle QSTR (Khadidja, 2015) 3
Figure 2. Des descripteurs
moléculaires relient la structure chimique à l'activité
biologique. 4
Figure 3. Exemple de tableaux de
connectivité C et de distance D de la molécule de l`acridine
7
Figure 4. Exemple d'illustration
d'échafaudage moléculaire 8
Figure 5. Illustration simplifiée d'un
réseau de neurones 10
Figure 6. L'arbre de décision a trois
types de noeuds 11
Figure 7. Cycle infectieux d'un coronavirus,
l'ensemble du processus de réplication virale a
lieu dans le cytoplasme 14
Figure 8. Illustration de la structure
moléculaire de l'amoxicilline 17
Figure 9. Illustration de la structure
moléculaire de l'azithromycine 17
Figure 10. Illustration de la structure
moléculaire des Fluoroquinolones 18
Figure 11. Illustration de la structure
moléculaire de Lopinavir (a) et Ritonavir (b) 19
Figure 12. Illustration de la structure
moléculaire du ribavirin 19
Figure 13. Illustration de la structure
moléculaire de favipiravid 20
Figure 14. Illustration de la structure
moléculaire de Remdesivir 20
Figure 15. Illustration de la structure
moléculaire de l'oseltamivir 21
Figure 16. Illustration de la structure
moléculaire de la chloroquine 21
Figure 17. Illustration de la structure
moléculaire des interférons 22
Figure 18. Illustration de la structure
moléculaire de la méthylprednisolone 23
Figure 19. Liste des herbes utilisées
par la médecine traditionnelle Chenoise. 24
Figure 20. Illustration de la plante «
Radix astragali » 24
Figure 21. Illustration de la plante «
Glycyrrhizae Radix Et Rhizoma » 25
Figure 22. Illustration de la plante «
Radix saposhnikoviae » 25
Figure 23. Illustration de la plante «
Rhizoma Atractylodis Macrocephalae » 26
Figure 24. Illustration de la plante «
Forsythiae Fructus » 26
Figure 25. Illustration du
chèvrefeuille et sa structure moléculaire 27
Figure 26. Illustration de la structure
moléculaire de digitoxigenin. 28
Figure 27. Illustration de la structure
moléculaire de Digitoxigenin en 3d. 28
Figure 28. Illustration de la structure
moléculaire de f3-Eudesmol. 29
Figure 29. Illustration de la structure
moléculaire de f3-Eudesmol en 3d. 29
Figure 30. Illustration de la structure
moléculaire de Crocin. 30
Figure 31. Illustration de la structure
moléculaire de Crocin en 3d. 30
Liste des tableaux
Tableau 1: L'interaction la plus importante de
la liaison hydrogène entre les trois composés
naturels et la protéine de pointe du coronavirus
(2019-nCoV). 28
1
Introduction générale
La découverte de nouveaux médicaments est
devenue assez important due à l'apparition fréquente de nouveaux
challenges liés avec la propagation de nouvelles épidémies
inattendues. Les dernières années, le monde a connu l'apparition
de plusieurs épidémies avec de virus plus dangereux
(l'épidémie avec MERS-CoV en 2012 (de Groot, 2013), d'Ebola en
2014 (Leroy, 2005), de SARS-COV 2 en 2019 (Van Doremalen, 2020), etc....).
Plus récemment, le monde a connu une propagation
sévère et rapide de l'épidémie causé par le
virus SARS-CoV-2 depuis la fin de l'année 2019 jusqu'à
maintenant. L'apparition de SARS-CoV-2 a été localisée
dans la ville de Wuhan, province du Hubei, Chine (Yang, 2020). Le 7 janvier
2020, il a été confirmé plus tard qu'il s'agissait d'un
nouveau type de coronavirus nommé SARS-CoV-2 (anciennement appelé
2019-nCoV). Cet événement inattendu a déclenché un
état d'alerte dans le monde entier. Cela a poussé les chercheurs
d'amplifier les efforts le plus tôt possible car il devenu très
urgent à réagir puisque ce virus a la possibilité de se
propager très rapidement et de tuer un grand nombre de gens.
La problématique essentielle réside au niveau de
l'efficacité d'une ou plusieurs approches à identifier des
médicaments contre le virus "SARS-CoV-2" dans un temps plus raisonnable
surtout que ce virus présente un danger très élevé
due à sa possibilité de se propager plus rapidement dans le
monde.
Notre objectif principal dans ce travail est l'identification
d'approches capable à répondre sur la problématique dans
un temps raisonnable et rendre ce document plus convaincant à donner une
idée claire sur l'efficacité de chaque approche à
répondre à la problématique et aussi nous visant que notre
travail puisse laisser une conclusion plus claire chez les lecteurs en cas
où nous parlons d'une comparaison entre les différentes approches
en termes d'efficacité.
Notre rapport documente les méthodes et approches les
plus adoptées par les chercheurs pour résoudre la
problématique principale qui est la recherches et la conception de
nouveaux médicaments de SARS-CoV-2. Le premier chapitre intitulé
"Modélisation QSPR/QSAR" est consacré, à une
présentation détailles de ces méthodes en introduisant
différentes étapes définissant la démarche
fondamentale de ces méthodes et leurs manipulation. Dans le second
chapitre intitulé «Conception et développement de nouveaux
médicaments contre le virus " SARS-CoV-2"», nous passons à
une étape plus profonde dans la révision générale
d'approches les plus convenable à réponse à la
problématique et aussi identifier celles qui ont réussi à
répondre en introduisant de médicaments capable de
prévenir le virus " SARS-CoV-2 dans un temps acceptable.
2
CHAPITRE 1 : Modélisation QSPR/QSAR
1. Les méthodes QSAR/QSPR
Les méthodes QSAR (Quantitative Structure-Activity
Relationships) et QSPR (Quantitative Structure Property Relationships) reposent
sur la recherche d'une relation entre un ensemble de nombres réels,
appelés descripteurs moléculaires et la propriété
ou l'activité que l'on souhaite prédire, afin de justifier les
données expérimentales disponibles et prédire les
propriétés et les activités pour des nouveaux
composés, pour lesquels les données expérimentales ne sont
pas disponibles (Denis, 2007). Le principe de ces dernières est
fondé sur l'outil statistique. En effet, il existe plusieurs types
différents d'outils statistiques :
> Régressions linéaires simples et multiples.
> Régressions aux moindres carrées partielles. > Arbres de
décision.
> Réseaux de neurones.
> Algorithmes génétiques.
> Vecteurs Machines.
Ces méthodes peuvent être utilisées pour
développer des modèles RQSA dans différent domaines
d'application comme la pharmacodynamique, pharmacocinétique et
toxicologie (Yap, 2005). Dernièrement, d'autres méthodes ont
été apparues comme les nouvelles méthodes basées
sur l'apprentissage automatique appelé « Machine learning »
qui permettent une modélisation plus complexe. Ces méthodes
surpassent fréquemment ceux développés à l'aide de
méthodes statistiques traditionnelles. Le modèle optimal est
obtenu en recherchant simultanément les paramètres de
modélisation optimaux et le sous-ensemble de caractéristiques. Ce
modèle sélectionné est vérifié avec les
paramètres optimaux en faisant une validation avec un ensemble de tests
pour s'assurer que le modèle est approprié et utile
(Figure 1).
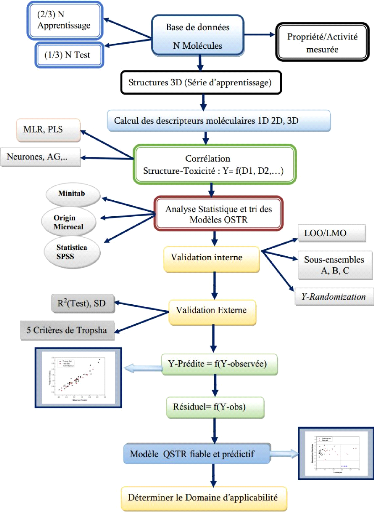
3
Figure 1. Schéma d'élaboration et
validation d'un modèle QSTR (Khadidja, 2015)
4
2. Méthodologie générale d'une
étude QSAR/QSPR
La qualité des résultats obtenus en suivant le
modèle QSAR dépendent des données expérimentales de
référence. Le choix de la base de données est une
étape très importante pour établir un lien avec les
données expérimentales de référence afin
d'éviter toutes erreurs probables. En se basant sur les données
expérimentales issues de la littérature, les théoriciens
peuvent donc choisir des données présentant des incertitudes
faibles et acceptables, ainsi ils auront des paramètres bien
ajustés aux données expérimentales. Les données
rassemblées doivent être obtenues en suivant un protocole
expérimental unique (Samir 2017).
2.1 Les descripteurs moléculaires
2.1.1 Définition
Les descripteurs moléculaires servent à extraire
et définir un ensemble d'informations concernant les
caractéristiques des molécules. Ils sont liés
implicitement ou explicitement aux
propriétés physico-chimiques de ces
dernières. Les informations liées avec
ces
caractéristiques seront traduites ensuite en une série de
grandeurs (en général scalaires) (Dudek, 2006). Ces grandeurs
sont appelés des descripteurs et sont déterminés pour
chaque molécule et ensuite liées mathématiquement à
l'activité biologique mesurée (Figure 2) (Kier,
1975). Un descripteur peut être parfois plus complexe comme par exemple
en cas de champs d'interaction dans l'espace 3D. Dans ce dernier cas, les
molécules sont superposées et alignées sur une grille et
les potentiels d'interaction sont déterminés pour chaque
molécule au niveau de chaque point de grille (CONSONNI, 2002).
En d'autres termes, les descripteurs sont nombreux et de
différentes complexités et de conceptions diverses.
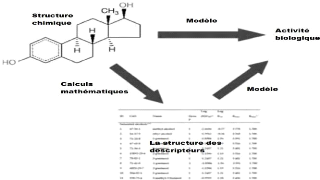
Figure 2. Des descripteurs moléculaires relient
la structure chimique à l'activité
biologique.
Le modèle QSAR/QSPR est une fonction
mathématique qui a comme paramètres les descripteurs
moléculaires et un objectif principal qui consiste à
déterminer la fonction biologique d'un produit chimique à partir
de sa structure.
5
2.1.2 Types de descripteurs
Dans la littérature, de nombreux descripteurs
moléculaires ont été introduits, plus de 10 000
descripteurs moléculaires qui quantifient des caractéristiques
physico-chimiques ou structurelles de molécules (Karelson, 2002). En
générale, ces descripteurs peuvent être obtenus par deux
façons différentes : empirique ou non-empirique, mais les
descripteurs calculés et non mesurés sont les plus
préférés, car ils répondent bien aux objectifs de
la modélisation puisque ils permettent des prédictions sans
passer par une étape de synthèse. Au contraire, il y a quelques
descripteurs mesurés qui utilisent généralement des
données expérimentales (polarisabilité, ou potentiel
d`ionisation...). La classification des descripteurs se fait fréquemment
en fonction de la dimensionnalité de la représentation
moléculaire sur laquelle ils sont calculés: on parlera donc de
descripteurs 0D, 1D, 2D et 3D. Malgré leur origine et leur
représentation mathématique différentes, la plupart des
descripteurs moléculaires sont interconnectés. Par exemple, les
changements topologiques des molécules peuvent être liés
aux changements de leur géométrie. Cependant, les changements au
niveau de la symétrie ou de la ramification peuvent affecter la
distribution de la charge électronique, ce qui peut causer des
changements dans la réactivité chimique ou la polarité,
respectivement. De même, dans certains cas, un paramètre
décrivant la distribution de la densité électronique au
sein de la molécule pourrait agir comme une meilleure mesure de
ramification, et donc de symétrie et de forme, ce qui est
irréalisable par les descripteurs topologiques typiques (Bauer,
1988).
a. Les descripteurs 0D
Tous les descripteurs moléculaires de cette classe 0D
ne nécessitent aucune information sur la structure moléculaire.
Les nombres d'atomes et de liaisons, ainsi que la somme ou la moyenne des
propriétés atomiques sont typiques de cette classe de
descripteurs. Ces descripteurs sont calculés, interprétés
facilement et ne nécessitent pas d'optimisation de la structure
moléculaire. Ils montrent généralement une très
forte dégénérescence, c'est-à-dire qu'ils ont des
valeurs égales pour plusieurs molécules, telles que les
isomères. Malgré que leur contenu informationnel est faible, mais
ils peuvent néanmoins jouer un rôle important dans la
modélisation de plusieurs propriétés physico-chimiques ou
prendre part à des modèles plus complexes.
b. Les descripteurs 1D
Cette catégorie de descripteurs est calculée
à partir de la formule brute de la molécule en utilisant la
composition moléculaire. Celle-ci nous permet de faire des calculs en
fonction des propriétés atomique de la molécule telles que
: les pourcentages massiques des atomes, la masse molaire, le poids
moléculaire etc. Il est noté MW et mesuré en daltons (Da).
Le poids molécule est définit sous forme de la somme des poids
atomiques des différents atomes constituant la molécule. Il est
utilisé dans l'étude de transport dont la diffusion et le mode de
fonctionnement. Plus les composés sont munis de poids
moléculaire, plus ils sont moins susceptibles d'être
absorbés. En effet, le fait de garder des poids moléculaires plus
bas que possible devrait être l'objectif pour établir un
médicament (Rekkab, 2014). Pour les médicaments
délivrés par voie orale le poids moléculaire doit
être inférieur ou égal à 500 daltons (optimum autour
de 300 daltons) (C.A. Lipinski, 1997).
6
Le pourcentage massique est défini par la formule
suivante : %massique =
Ces descripteurs montrent généralement une
dégénérescence moyenne plus élevée,
cependant ils sont très utiles dans la modélisation à la
fois des propriétés physico-chimiques et biologiques.
c. Les descripteurs 2D
Les descripteurs moléculaires 2D utilisent une
représentation sous forme de graphes dits «descripteurs 2D»
(ou indices topologiques). Dans cette catégorie on trouve principalement
les descripteurs topologiques qui contiennent des informations relatives
à la connectivité ainsi que des estimations des
propriétés physicochimiques. Ce sont des descripteurs plus riches
d'information qui permettent la prédiction de la majorité des
propriétés moléculaires. Les informations sur les
propriétés moléculaires peuvent inclure Le nombre de
liaisons, les mesures de branchement et les représentations
théoriques des graphes de la structure moléculaire sont des
descripteurs courants de ce type. Les descripteurs 2D sont
considérés un peu plus complexes en comparaison avec les
descripteurs 0D ou 1D. Ces descripteurs 2D sont généralement
calculés en ensembles et sont représentés sous forme de
tableaux (Figure 3) de nombres binaires, entiers ou
réels (Euler, 1976) et (Schultz, 1989). En se basant sur ces tableaux,
nous pouvons donc calculer de nombreux indices topologiques. Parmi ceux, que
nous pouvons calculer, on trouve par exemple:
? L`indice de Wiener (Wiener, 1947), cet
indice permet de calculer le nombre total de liaisons dans les chemins les plus
courts entre toutes les paires d`atomes (en excluant les hydrogènes). Il
est défini par la formule suivante :
?
Avec : N est le nombre des atomes de la molécule et est
le plus petit nombre de liaisons séparant les deux atomes i et j.
? L'indice de Balaban (Balaban, 1982),
noté (J), est l`un des indices topologiques les plus importants et qui
permet de décrire le degré de ramification des molécules
non cycliques. Il est défini par la formule suivante :
?( )
Avec : i et j sont les atomes voisins (autres que
l`hydrogène), N est le nombre d`atomes
de la molécule , et sont les connectivités des
atomes i et j, est le nombre des liaisons,
et est le nombre des cycles.
? La somme des degrés de valence (D.
Jaiswal, 2006) , notée (SVD), est définit sous forme de la somme
de tous les degrés de valence de la molécule
représentée par un graphe, le degré d`un point
correspondant au nombre de lignes se terminant par ce point. Ce
paramètre dépend donc principalement de la ramification de la
molécule.

7
Figure 3. Exemple de tableaux de
connectivité C et de distance D de la molécule de l`acridine
d. Les descripteurs 3D
Les descripteurs moléculaires 3D sont basés sur
l'utilisation des positions relatives des atomes dans l'espace. Ils peuvent
décrire des caractéristiques plus complexes ; leurs calculs
nécessitent donc de connaître la géométrie 3D de la
molécule en passant le plus souvent par une modélisation
moléculaire empirique ou ab-initio. Ces descripteurs présentent
un temps de calcul relativement coûteux, mais apportent davantage en
termes d'informations fournis. Ils sont nécessaires à la
modélisation de propriétés ou d'activités qui
dépendent de la structure 3D. On distingue plusieurs familles
importantes dans cette catégorie de descripteurs.
* Les descripteurs géométriques:
les plus populaires sont le volume moléculaire, la surface
accessible au solvant et le moment principal d'inertie.
* Les descripteurs électroniques:
permettent de quantifier différents types d'interactions inter-
et intramoléculaires liées à l'activité biologique
des molécules. Le calcul de la plupart
de ces descripteurs nécessite la recherche de la
géométrie pour laquelle l'énergie stérique est
minimale, et cela se fait souvent à l'aide de la chimie quantique. Par
exemple, les énergies de la plus haute orbitale moléculaire
occupée (HOMO) et de la plus basse vacante (LUMO) (orbitales
frontières) sont des descripteurs fréquemment
sélectionnés. Le moment dipolaire, le potentiel d'ionisation et
différentes énergies relatives à la molécule sont
d'autres paramètres importants.
* Les descripteurs spectroscopiques: en
utilisant ces descripteurs, les molécules peuvent être
caractérisées par des mesures spectroscopiques en fonction de
leurs ondes vibrationnelles. En effet, les vibrations d'une molécule
dépendent de la masse des atomes et des forces d'interaction entre
ceux-ci; ces vibrations fournissent donc des informations sur la structure de
la molécule et sur sa conformation. Les spectres infrarouges peuvent
être obtenus soit de manière expérimentale, soit par un
calcul théorique, après recherche de la géométrie
optimale de la molécule
e. Les descripteurs locaux des
propriétés surfaciques moléculaires
Ce descripteur a été introduit par les
chercheurs en se basant sur le principe d'échafaudage moléculaire
« Scaffold ». Ce principe est l'un des concepts les plus importants
et les plus largement utilisés en chimie médicinale. Il utilise
les propriétés moléculaires locales pour extraire des
informations à partir de leur nature surfacique et structurelle de
constitution (Figure 4). L'échafaudage
représente généralement les structures centrales du cadre
moléculaire. Dans la littérature, plusieurs définitions
ont été introduites à propos des échafaudages
moléculaires, mais la définition la plus largement adoptée
est donnée par (Bemis, et al., 1996), en obtenant l'échafaudage
par la suppression de toutes les chaînes latérales (ou groupes R).
Parmi tous les échafaudages introduits dans littérature, dans ce
chapitre, on s'intéresse plus spécifiquement à ceux ayant
des propriétés de bioactivité
préférées (qui sont appelés "échafaudages
privilégiés" (Barreiro, London, 2015) ou "échafaudages
bioactifs" (Varin, et al., 2011) et (Nakagawa, et al., 2018)) qui bien
sûr présentent un intérêt particulier pour la
découverte de médicaments. De plus, plusieurs études ont
démontré que ces échafaudages sont pertinents pour la
chimie computationnelle et médicinale. Par exemple, l'analyse
informatique vise à isoler et comparer systématiquement les
structures centrales des composés actifs.
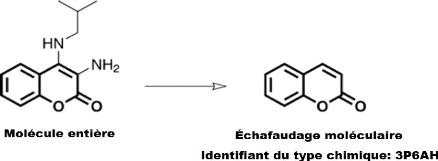
8
Figure 4. Exemple d'illustration d'échafaudage
moléculaire
f. Les descripteurs
quantiques/électroniques
Ces descripteurs s'intéressent à des
caractéristiques supplémentaires de la structure
moléculaire, surtout les informations moléculaires liées
avec la densité électronique résultante de distribution de
charge des molécules et ceux liées avec la chimie quantique sous
forme de données structurales, énergétiques,
électroniques et spectroscopiques (Mezey, 1993) et (Mezey, 1999) .
L'analyse de la densité électronique nous permet donc de
quantifier les différents types d'interactions inter et
intramoléculaires qui ont une relation potentille avec l'activité
biologique au niveau de la molécule.
2.2 Méthodes d'analyse de données
L'objectif principal des méthodes d'analyse de
données est d'utiliser la statistique pour extraire des informations
utiles des données à fin d'analyser au mieux l'incertitude et
la
variation dans les observations. L'analyse de données
est plus importante pour décrire, comprendre, évaluer
suffisamment les phénomènes étudiés et
d'interpréter les résultats trouvés d'une façon
claire. Les données peuvent être de toute nature, ce qui rend la
statistique très utile dans la plupart des disciplines :
économie, sociologie, psychologie, agronomie, biologie, médecine,
chimie, physique, géologie, sciences de l`ingénieur, sciences de
l`information et de la communication, etc. (ROY, 2015). La mise en place des
modèles QSAR/QSPR à l'aide des méthodes statistiques n'est
pas une tâche facile à réaliser due à la
difficulté au niveau de la différence d'échelles existant
entre les données à corréler. Par exemple, la structure
étant à une échelle moléculaire alors que les
propriétés à prédire sont à une
échelle macroscopique. De l'autre côté, on doit tenir
compte des problèmes d'incertitude à la fois au niveau des
structures moléculaires (liées niveau de calcul) et des
données expérimentales (protocoles de mesures). De plus, le
traitement d'une grande quantité de données génère
une difficulté supplémentaire lié avec le processus de
mise au point de modèles QSAR, surtout quand on veut analyser les
corrélations entre un grand nombre de descripteurs d'un grand nombre de
molécules ce qui entraine une perturbation au niveau du choix des
paramètres structuraux parmi ceux disponibles. Dans la
littérature, il existe de nombreuses méthodes d'analyse de
données, mais dans ce chapitre nous allons s'intéresser aux
quelques méthodes les plus utilisées.
2.2.1 Méthodes basées sur les descripteurs
Les méthodes statistiques basées sur les
descripteurs, permettent de représenter numériquement la
structure chimique pour en déduire ensuite un modèle. La mise en
pratique de cette méthode dans la recherche de nouveaux
médicaments peut bien refléter l'importance de cette
méthode. Une application de cette méthodologie est de pouvoir
calculer par exemple la capacité d'une molécule d'être un
candidat médicament ou une tête de série afin de
réduire les risques d'échecs aux étapes
expérimentales.
a. Approches linéaires
En général, les fonctions linéaires sont
facilement interprétables et suffisamment précises pour de
petites séries de composés identiques, spécialement
lorsque les descripteurs sont sélectionnés avec soin pour une
grandeur donnée.
b. La régression linéaire multiple
(MLR)
La régression linéaire multiple MLR est l'une
des méthodes de modélisation les plus populaires grâce
à sa simplicité d'utilisation et facilité
d`interprétation. L'avantage important de la régression
linéaire multiple est qu'elle est très transparente, puisque
l'algorithme est disponible, et que les prédictions peuvent être
réalisées facilement (Fernández, 2007). L'analyse de
régression linéaire multiple repose sur l'hypothèse qu'il
existe une relation linéaire entre une variable dépendante Y et
une série de n variables indépendantes Xi. Pour les études
de régression multiple, le nombre de variables doit être
inférieur ou égal au nombre d'individus (molécules).
L'objectif est d'obtenir une équation de la forme suivante :
9
X1,...., Xn sont des descripteurs moléculaires
affectés de leurs coefficients a1,....an.
Les coefficients ai peuvent être obtenus en utilisant
des estimateurs comme la méthode des moindres carrés qui minimise
la somme des résidus au carré. Les valeurs des coefficients peut
exprimer le degré d'influence des descripteurs moléculaires
utilisés sur la propriété cible. De plus, un coefficient
positif indique que le descripteur moléculaire correspondant contribue
positivement à la propriété cible, tandis qu'un
coefficient négatif indique une contribution négative.
c. La méthode de régression des moindres
carrés partiels
La régression par les moindres carrés partiels
(PLS) est une technique qui sert à optimiser les calculs en diminuant le
nombre de descripteurs à un plus petit ensemble de composantes non
corrélées et effectuer la régression par les moindres
carrés sur ces composantes, plutôt que sur les données
initiales (Hasegawa, 2010). L'analyse avec la méthode PLS fournit des
résultats avec moins d'incertitude des mesures.
d. Approches non linéaires
Les méthodes non-linéaires étendent les
calculs avec l'approche QSAR à des relations plus complexes. Ces
méthodes souffrent des difficultés et parfois sont
sur-ajustés (ils se borneront dans ce cas à décrire du
bruit au lieu de la relation sous-jacente entre descripteurs et
activité). Malgré ces inconvénients, la recherche
pharmaceutique tire un grand bénéfice de l'application des
méthodes non linéaires.
e. Réseaux de neurones artificiels
La méthode des réseaux de neurones artificiels
sont des modèles mathématiques qui suivent le même principe
que le cerveau humain, mais d'une façon plus simplifiée. Les
réseaux de neurones sont des systèmes de traitement de
l'information basés sur des outils mathématiques et
algorithmiques qui s'avèrent être puissants et commodes pour
résoudre des problèmes complexes (Breneman, 2003). Un
réseau de neurones est un processeur massivement distribué en
parallèle qui a une propension naturelle pour stocker de la connaissance
empirique et la rendre disponible à l'usage. Il ressemble au cerveau
humain sur deux aspects :
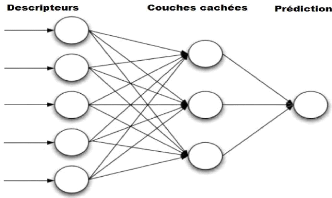
Figure 5. Illustration simplifiée d'un
réseau de neurones
10
11
Les réseaux de neurones sont basés sur trois
couches: la couche d'entrée des neurones, au moins une couche
cachée de neurones et une couche de sortie des neurones (Figure
5). Ces réseaux peuvent utiliser de couches
supplémentaires de neurones en cas de complexité
élevée pour capturer des informations plus précises
concernant les relations moléculaires. Ils sont formés de
manière itérative, où chaque période de formation
est appelée une époque. Ainsi qu'une une phase
d'entraînement est un processus itératif sert à la
minimisation de l'erreur entre l'activité connue et l'activité
prédite par le réseau neuronal.
f. Arbre de décision
L'arbre de décision est un concept utilisé dans
la théorie des graphes, un arbre est un graphe non orienté,
acyclique et connexe. L'ensemble des noeuds se divise en trois
catégories :
? Noeud racine (l'accès à l'arbre se fait par ce
noeud).
? Noeuds internes : les noeuds qui ont des descendants, qui sont
à leur tour des noeuds. ? Noeuds terminaux (ou feuilles) : noeuds qui
n'ont pas de descendant.
Un arbre de décision est un schéma qui
représente les résultats possibles d'une série de choix
interconnectés. Il permet à une personne ou une organisation
d'évaluer différentes actions possibles en fonction de leur
coût, leur probabilité et leurs bénéfices. Il peut
être utilisé pour
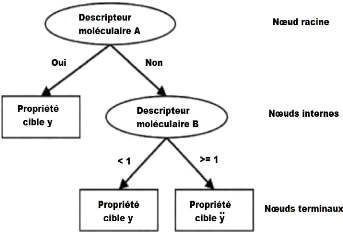
Figure 6. L'arbre de décision a trois types de
noeuds
exploiter des relations entre les données en permettant
de décrire ces données en se basant sur une combinaison de
techniques mathématiques et de calcul pour faciliter la description, la
catégorisation et la généralisation d'un ensemble de
données.
12
2.3 Interprétation et validation d'un modèle
QSPR/QSAR
La validation du modèle QSPR/QSAR se fait après
avoir établi un certain ensemble d'étapes de
développement. Ces étapes importent la définition de la
base des données, le choix et le calcul des descripteurs, établir
la corrélation entre différents descripteurs et analyse
statistique. Une fois ces étapes sont faites, le modèle doit
être interprété en analysant tous les paramètres
statistiques de ce modèle en se basant sur la méthode d'analyse
adoptée, sa qualité doit être bien étudiée,
cette qualité est vérifiée pour une validation. Sa
robustesse, c'est-à-dire l'influence des composés de la
série d'apprentissage sur le modèle est estimée par des
méthodes de validation interne. Afin d'estimer son pouvoir
prédictif, des données expérimentales
supplémentaires sont nécessaires afin de déterminer la
capacité du modèle à prédire ces valeurs c'est ce
que l'on appelle validation externe. Enfin, il est important de savoir quel
type de molécules utilisées avec quel modèle. On parle
alors de domaine d'applicabilité.
2.3.1 Validation interne
Dans le passé, la validation interne d'un modèle
QSPR/QSAR a été réalisée en utilisant la validation
croisée LOO (Leave One Out) ou LMO (Leave Many Out) qui est
quantifiée par le
coefficient (L. Zhang, 2008). Ce processus consiste à
extraire un certain nombre k de
molécules du jeu initial à N
molécules et à construire un nouveau modèle avec les (N-k)
molécules restantes à l'aide des descripteurs choisis (seules les
constantes de la régression changent). Ce processus est ensuite
réitéré pour retirer et prédire les valeurs de
toutes les molécules de la série d'apprentissage. En fonction du
nombre de molécules retirées à chaque itération, on
parlera de LOO ou de LMO selon qu'une ou plusieurs molécules est (sont)
retirée(s). Dans ces dernières années, d'autres
méthodes sont utilisées pour faire la validation interne, tel que
la hasardisation de la réponse (Y-Randomization) (L. He, 2005).
Cependant, la validation interne a montré son insuffisance pour
étudier le pouvoir prédictif d'un modèle QSPR/QSAR. Cette
insuffisance de prédiction a permet l'adoption de la validation externe
du modèle comme une norme et une partie obligatoire dans la
modélisation basée sur les méthodes QSPR/QSAR (Tropsha,
2003).
2.3.2 Validation externe
En arrivant à l'étape de validation par cette
méthode, notre objectif principal est de prédire la
propriété et l'activité d'une série de
molécules appelée généralement série de
test. La validation externe doit être effectué dans le cadre d'une
prévision des composés issus d'un ensemble n'ayant pas
été utilisé dans l'élaboration du modèle.
Elle est caractérisée par les
paramètres (test), (test). Récemment plusieurs
études (Golbraikh, 2002) ont montré
l'insuffisance de ces
paramètres pour vérifier le pouvoir prédictif des
modèles QSAR et QSPR. Par conséquent, d'autres paramètres
doivent être vérifiés pour cet objectif. Ces
paramètres sont connus sous le nom « critères de validation
externe » ou souvent appelés « critères de Trophsa
».
3. 13
Applications
Les applications des méthodes QSPR/QSAR sont multiples
à savoir la prédiction des propriétés
physico-chimiques, activités biologiques, etc.
Propriété physico-chimiques :
> Point d'ébullition, point de fusion, densité,
température critique.
> Solubilité.
> Pression de vapeur ...
Activités biologique :
> Anti VIH.
> Anti cancer.
> Anti Covid sars.
> Anti Malaria.
> Anti inflammatoire ...
Autres propriétés/activités :
> Prédiction de la toxicité aquatique des
composés chimiques.
> Toxicité des nanoparticules.
> Toxicité des pesticides et des colorants.
> Concentration micellaire critique.
> Propriétés inhibitrices de corrosion.
> Conception des médicaments et de nombreux autres
produits tel que les parfums ;
les colorants et les produits de la chimie fine ...
4. Conclusion
Dans le premier chapitre, nous avons s'intéressé
à la modélisation avec les méthodes QSAR/QSPR et la
classification de différents descripteurs utilisés pour exprimer
l'activité biologique de manière quantitative et le passage
à des expressions mathématiques qui peuvent utilisés comme
moyen prédictif de la réponse biologique. Dans le chapitre
suivant, nous révisons les approches adoptées dans la recherche
de nouveaux médicaments de "SARS-CoV-2".
14
CHAPITRE 2 : Conception et
développement de nouveaux médicaments
Contre le virus " SARS-CoV-2"
1. Introduction
Ces derniers temps, il y a eu une propagation d'une pneumonie
inattendue dans la ville de Wuhan, province du Hubei, La chine (Yang, 2020). Le
7 janvier 2020, il a été confirmé plus tard qu'il
s'agissait d'un nouveau type de coronavirus nommé SARS-CoV-2.
L'apparition de ce nouveau virus a poussé l'organisation mondiale de la
santé (OMS) à l'appeler la pneumonie de Wuhan en tant que maladie
à coronavirus-2019 (COVID-19) le 11 février 2020 (Yang, 2020).
Delors, le virus de COVID-19, fait l'objet de plusieurs recherches
scientifiques autour du monde entier. Cette révolution contre COVID-19
est considérée comme une réaction émergente
naturelle, car ce virus a des effets rapides et dangereux sur l'homme et sur
son économie. Les effets liés à la contamination par ce
virus comprennent des symptômes respiratoires (toux, fièvre et
lésions pulmonaires ...) et certains autres symptômes tels que
fatigue, myalgie et diarrhée ( (GUAN, 2020), (HUANG, 2020)). Les
coronavirus sont une grande famille de virus qui provoquent des maladies allant
du simple rhume à des maladies plus compliquées, en effet ce
virus peut subir des mutations par le changement de l'ARN pour devenir un autre
organisme, lorsque le virus COV-19 pénètre dans la cellule ;
l'opération se termine par la mort de cette cellule (Figure
7).
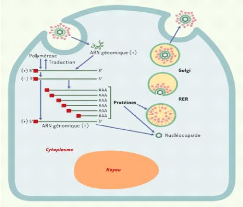
Figure 7. Cycle infectieux d'un
coronavirus, l'ensemble du processus de réplication
virale a lieu
dans le cytoplasme
15
La propagation rapide est également
considérée comme un défi difficile à relever. Les
chercheurs et l'organisation mondiale de la santé ont expliqué
cette propagation rapide de Covid-19 pour certaines raisons telles que la
transmission interhumaine, la capacité du virus à rester
attaché plusieurs aux surface du plastique, bois, papiers et autres, ce
qui peut entraîner une augmentation du taux de propagation du virus par
la transmission interhumaine lorsque quelqu'un touche des endroits
épidémiques. La capacité du virus à s'attacher aux
matières dépend de chaque type de matière, par exemple, il
reste attaché jusqu'à 7 jours aux plastiques et trois heures sur
les papiers. Afin de stopper la propagation du coronavirus, plusieurs
programmes de prévention ont été mis à travers le
monde à titre d'exemple le confinement qui a pu donner un coup de frein
contre ce virus impitoyable en attendant l'élaboration du vaccin par les
virologues.
2. Les approches adoptées dans la recherche de
nouveaux médicaments de "SARS-CoV-2".
Récemment, diverses approches sont adoptées pour
rechercher de nouveaux médicaments de "SARS-CoV-2". Ces approches
peuvent inclure des approches informatiques proposées pour la conception
du médicament et de vaccin (Kumar, 2020), approches basées sur la
médecine traditionnelle, les fragments, la carte de connectivité
(Connectivity Map) et la médecine in silico.
2.1 Approche basée sur la médecine
traditionnelle
La médecine traditionnelle est connue au monde entier
sous le nom de médecine indigène ou populaire. Ce type de
médecine comprend plusieurs techniques et aspects médicaux des
connaissances traditionnelles accumulées depuis l'antiquité et
qui se sont développées d'une génération à
une autre au sein de diverses sociétés avant la naissance de la
médecine moderne. L'organisation mondiale de la santé (OMS)
définit la médecine traditionnelle comme «l'ensemble des
connaissances, des compétences et des pratiques fondées sur les
théories, les croyances et les expériences propres à
différentes cultures, explicables ou non, utilisées
également pour le maintien de la santé. Parmi d'eux il y'a la
Médecine traditionnelle chinoise (CHEN, 2011), cette médecine
vieille d'au moins 23 siècles vise à continuer sa mission dans le
monde actuel, l'acupuncture et les remèdes à base de plantes
chinoises remontent à au moins 2200 ans. Les guérisseurs chinois
traditionnels cherchent à rétablir un équilibre dynamique
entre deux forces complémentaires, le yin (passif) et le yang (actif),
qui imprègnent le corps humain. Selon elle, une personne est en bonne
santé lorsqu'il existe une harmonie entre ces deux forces; la maladie,
en revanche, résulte d'une rupture de l'équilibre du yin et du
yang.
2.2 Approche basée sur les fragments
Les approches basées sur les fragments visent à
trouver de nouvelles petites molécules qui se lient aux protéines
dans le contexte la biologie chimique (Scott, 2012). Les résultats
obtenus sont en général très encourageantes aux
chercheurs. La méthodologie de fragments est initialement
développée dans quelques centres de l'industrie biotechnologique
et pharmaceutique, cette méthodologie a maintenant été
largement adoptée à la fois dans l'industrie pharmaceutique et
dans le monde universitaire. Après le succès initial avec des
16
cibles de kinases, la polyvalence de cette approche s'est
maintenant étendue à une large gamme de différentes
classes de protéines. Le rôle des approches basées sur les
fragments dans un environnement de recherche universitaire est également
montrer la capacité de cette méthodologie à aider les
chercheurs à mettre l'accent sur les maladies négligées
telles que la tuberculose. Le développement d'une bibliothèque de
fragments aide à faire un criblage de fragments, ce qui présente
donc un avantage tout à la fois pratique et théorique.
2.3 Approche basée sur la carte de
connectivité (Connectivity Map)
Cette technologie consiste à établir un profil
d'expression à l'échelle du génome des transcrits de
gènes (Qu, 2012). Cette possibilité d'expression a
été appliquée avec succès dans la découverte
biomédicale pendant plus d'une décennie. Basé sur sa
validité scientifique par rapport aux résultats
expérimentaux de cette technologie, Connectivity Map fournit une
approche systématique basée sur la recherche des relations entre
les gènes, les produits chimiques et les conditions biologiques. Depuis
sa première introduction en 2006, cet approche a montré de
nouvelles promesses dans le domaine de développement de
médicaments, telles que l'identification et la suggestion de nouvelles
indications pour les médicaments existants et l'élucidation du
mode d'action pour de nouveaux produits chimiques en plus de prédire
potentiellement les effets secondaires.
2.4 Approche basée sur la médecine in
silico
La médecine in silico (également connue sous le
nom de "médecine computationnelle") est l'application de la recherche in
silico aux problèmes concernant la santé et la médecine
(ZHAVORONKOV, 2020). C'est l'utilisation directe de la simulation informatique
dans le diagnostic, le traitement ou la prévention d'une maladie. Plus
précisément, la médecine in silico se caractérise
par la modélisation, la simulation et la visualisation de processus
biologiques et médicaux dans les ordinateurs dans le but de simuler de
vrais processus biologiques dans un environnement virtuel (Mak, 2019).
3. Identification de médicaments antibiotiques de
"SARS-CoV-2" en l'absence d'une thérapeutique antivirale et d'un vaccin
spécifiques
3.1 Des médicaments antibiotiques
Les médicaments antibiotiques sont des
dérivés de substances isolées dans la nature. Certains
antibiotiques agissent sur la paroi ou la membrane des bactéries et
ainsi ils causent leur destruction. D'autres vont agir au niveau de la
machinerie des bactéries pour bloquer leur développement et ainsi
bloquer leur survie. Enfin, certains antibiotiques agissent directement au
niveau de leur ADN pour empêcher leur division et ainsi empêcher
leur réplication en plusieurs copies et leur prolifération.
3.1.1 Amoxicilline
L'amoxicilline est un antibiotique qui appartient au groupe
des aminopénicillines, de la famille des béta lactamines (Duval,
1973). Son mode d'action antibiotique aide à faire un blocage de la
synthèse des parois bactériennes. Certaines bactéries
sécrètent des enzymes, les béta-lactamases, qui inactivent
l'action de l'amoxicilline. Cette action peut être restaurée
par
17
l'association de l'amoxicilline à un inhibiteur des
béta-lactamases, l'acide clavulanique, qui annule l'action de ces
enzymes. La structure moléculaire de l'Amoxicillin est
représentée comme suit (Figure 8).
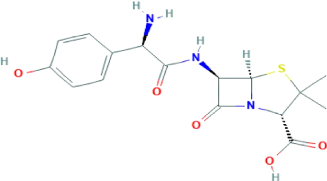
Figure 8. Illustration de la structure
moléculaire de l'amoxicilline
3.1.2 Azithromycine
L'Azithromycine est un médicament antibiotique de la
famille des "macrolides" commercialisé en France depuis 2008 (CLYTI,
2004). Elle agit essentiellement sur les bactéries atypiques notamment
celles avec un mécanisme intracellulaire. De façon
générale, les antibiotiques macrolides sont utilisés pour
traiter des infections des voies respiratoires (nez, gorge, bronches, poumons),
de la bouche, des oreilles, les infections cutanées et les infections
des organes génitaux. La structure moléculaire de l'Azithromycine
est représentée comme suit (Figure 9).
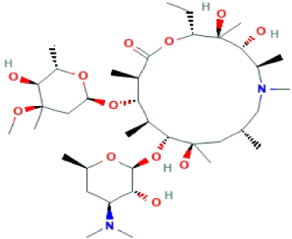
Figure 9. Illustration de la structure
moléculaire de l'azithromycine
18
3.1.3 Fluoroquinolones
Les Fluoroquinolones sont des antibiotiques
bactéricides qui se dérivent des quinolones par des modifications
chimiques (SMITH, 2001). Cette dérivation se termine notamment par
l'ajout d'un atome de fluor. Ces transformations chimiques donnent la naissance
des Fluoroquinolones avec une activité in vitro 100 fois
supérieure à celle des quinolones. Ces médicaments sont
des antibiotiques fréquemment utilisés en pratique courante et se
présentent sous forme de comprimés et de flacons injectables en
perfusion par voie intraveineuse. La structure moléculaire des
Fluoroquinolones est représentée comme suit (Figure
10).
Figure 10. Illustration de la structure
moléculaire des Fluoroquinolones
3.2 Des médicaments antiviraux
Les antiviraux sont des molécules qui ont pour but de
lutter contre des virus. Ils sont administrés sous forme de
médicaments pour lutter contre une infection virale. Ils empêchent
donc une infection virale de se répandre dans les cellules. Ils
n'éradiquent pas pour autant les virus.
3.2.1 Lopinavir / Ritonavir
Le lopinavir est un antirétroviral qui joue le
rôle d'inhibiteur de protéase. Ce médicament est largement
utilisé pour le traitement du VIH (Cvetkovic, 2003). Il est
formulé en association avec un autre inhibiteur de protéase, le
ritonavir (lopinavir / ritonavir, de marque Kaletra ou Aluvia). Le ritonavir
jeu un rôle assistant en inhibant l'enzyme métabolisante du
cytochrome P450 3A et augmente donc la demi-vie du lopinavir2. La structure
moléculaire du lopinavir et le ritonavir est représentée
comme suit (Figure 10).
19
Figure 11. Illustration de la structure
moléculaire de Lopinavir (a) et Ritonavir (b)
3.2.2 Ribavirin
Le Ribavirin est un médicament indiqué dans le
traitement de l'hépatite C chronique en association avec d'autres
médicaments (Gilbert, 1986). Cependant son utilisation
thérapeutique se limite essentiellement au traitement de
l'hépatite C chronique et plus rarement du virus syncytial respiratoire
(RSV). Les limites de son utilisation sont principalement liées à
des problèmes pharmacologiques de biodisponibilité et de
toxicité qu'à une activité antivirale insuffisante. La
structure moléculaire de Ribavirin est représentée comme
suit (Figure 12).
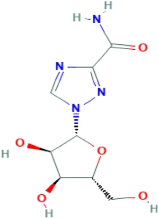
Figure 12. Illustration de la structure
moléculaire du ribavirin
3.2.3 Favipiravid (T-705)
Le médicament Favipiravir ou T-705, est une pyrazine
organofluorée utilisée comme antiviral contre les virus à
ARN, notamment les orthomyxovirus (dont les différents virus de la
grippe), le virus du Nil occidental, le virus de la fièvre jaune, le
virus de la fièvre aphteuse
ainsi que d'autres flavivirus, arénavirus, bunyavirus,
alphavirus2 (Furuta, 2013). La structure moléculaire de Favipiravir est
représentée comme suit (Figure 13).
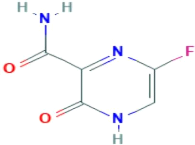
Figure 13. Illustration de la structure
moléculaire de favipiravid
3.2.4 Remdesivir
Remdesivir est un médicament expérimental
initialement développé pour soigner les malades de la
fièvre hémorragique Ebola (Wang, 2020). Ce médicament est
la première thérapie à avoir démontré une
certaine efficacité dans un essai aussi grand avec plus d'un millier de
patients. Les résultats de cet essai à été
considéré modeste. La structure moléculaire de Remdesivir
est représentée comme suit (Figure 14).
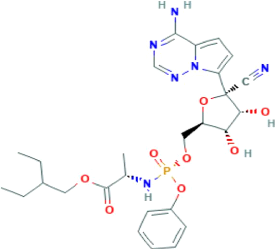
20
Figure 14. Illustration de la structure
moléculaire de Remdesivir
3.2.5 Oseltamivir
L'oseltamivir est un médicament antiviral
utilisé pour le traitement des grippes A et B. Ce médicament
réduit aussi les risques de complication (McClellan, 2001). Il est
produit à partir d'acide shikimique qui est un inhibiteur de la
neuraminidase, un enzyme qui est situé au niveau de la membrane du virus
et il est indispensable à la libération des virions à la
fin du cycle de réplication virale. La structure moléculaire
d'oseltamivir est représentée comme suit (Figure
15).
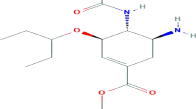
Figure 15. Illustration de la structure
moléculaire de l'oseltamivir
3.2.6 Chloroquine
La chloroquine est un antipaludique qui appartient à la
famille des 4-aminoquinoléines. Ce médicament a été
largement commercialisé sous forme de sels (sulfate ou phosphate) (Wang,
2020). Le traitement avec la chloroquine a été
considéré le plus efficace contre le paludisme, en
préventif comme en curatif. Elle est aussi très utilisée
contre des maladies auto-immunes telles que le lupus et des maladies
rhumatoïdes telles que la polyarthrite rhumatoïde. Elle montre in
vitro des effets antiviraux, mais qu'on n'arrive pas ou mal à reproduire
in vivo. La structure moléculaire de la chloroquine est
représentée comme suit (Figure 16).
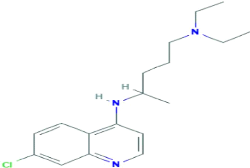
21
Figure 16. Illustration de la structure
moléculaire de la chloroquine
3.2.7 Interférons
Les interférons sont des glycoprotéines de la
famille des cytokines. Ces protéines sont naturellement produites par
les cellules du système immunitaire, mais également par d'autres
types cellulaires (cellules dendritiques, mononuclées,
épithéliales, etc.) (Isaacs, 1964). Ils sont produits en
réponse à la présence d'une double hélice d'ARN
étranger dans l'organisme. Le rôle principal des
interférons est de défendre l'organisme des agents
pathogènes tels les virus, bactéries, parasites et cellules
tumorales. Cela se fait en induisant la production de protéines de la
fonction immunitaire (notamment antivirales et anti-bactériennes, ou
à effet sur la réponse immune, et à viser
anti-prolifératives). La structure moléculaire des
interférons est représentée comme suit (Figure
17).
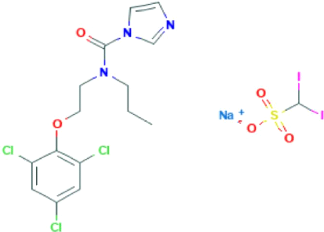
22
Figure 17. Illustration de la structure
moléculaire des interférons
3.3 Les corticostéroïdes
(méthylprednisolone)
Les corticostéroïdes sont utilisés pour
traiter des affections inflammatoires comme celles causées par l'asthme,
la polyarthrite rhumatoïde et aussi celle des maladies inflammatoires de
l'intestin. Ces médicaments peuvent être aussi utilisés
pour traiter les éruptions cutanées et des douleurs musculaires.
La méthylprednisolone ou le 6-alpha-méthylprednisolone, est un
corticoïde de la famille des glucocorticoïdes (comme la
prednisolone ou le cortisol) (STRUPP, 2004). Elle est
utilisée dans les traitements anti-inflammatoires (comme l'allergie)
dans les dérèglements du sang et les vomissements tardifs induits
par la chimiothérapie. La méthylprednisolone est la substance
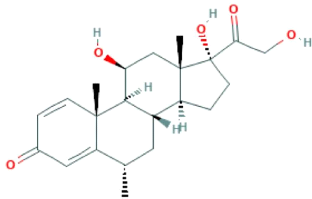
active du Médrol, du Solumédrol. La structure moléculaire
de la méthylprednisolone est représentée comme suit
(Figure _18).
23
Figure 18. Illustration de la structure
moléculaire de la méthylprednisolone
4. Identification de nouveaux médicaments herbal de
"SARS-CoV-2"
La plupart des médicaments herbals qui sont
identifiés convenables pour traiter le virus « SARS-CoV-2 »
sont des médicaments traditionnels chinois et japonais. Ces
médicaments ont été largement utilisé à la
fois en tant qu'herbe unique et dans les prescriptions de composés en
Asie, principalement en raison de ses effets de dissipation thermique et de
détoxication. La pharmacologie moderne a prouvé que chaque herbe
possède divers effets thérapeutiques, in vitro et in vivo, tels
que des activités anti-inflammatoires, antibactériennes et
antivirales. Dans cette partie du chapitre, nous intéressons à
l'identification de la structure moléculaire de chaque herbe qui a
montré son efficacité pour luter contre le virus SARS-CoV-2.
D'une autre part, la médecine traditionnelle Marocaine a aussi
montré sa capacité de lutter contre le virus SARS-CoV-2 via
l'utilisation de certains composés naturels qui existent au Maroc
à base de plantes médicinales qui reflètent de bon
résultats dans ce sens.
4.1 Les médicaments anti-coronavirus à base
de la médecine traditionnelle Chenoise
Dans cette section, nous allons introduire la liste des herbes
utilisés par la médecine traditionnelle Chenoise destinés
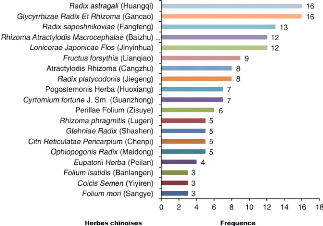
au traitement du SARS-CoV-2 (Yang, 2020). Les résultats ont
montré que ces formules contenaient 54 herbes différentes, dont
19 herbes avec une fréquence d'utilisation pendant 3 fois ou plus dans
les formules préventives pour la population générale
(Figure 19). Dans le reste de cette section, nous allons
introduire quelques principales plantes utilisées dans la
médecine traditionnelle Chenoise.
24
Figure 19. Liste des herbes
utilisées par la médecine traditionnelle Chenoise.
4.1.1 Radix astragali (Huangqi)
Radix Astragali (racine d'Astragale; Huangqi) (Figure
20) est très connue dans la médecine traditionnelle
chinoise courante (Ma, 2002). Cette herbe est avéré être un
immunostimulant,

Figure 20. Illustration de la plante « Radix
astragali »
tonique (adaptogène), hépatoprotecteur,
diurétique, antidiabétique, analgésique, expectorant et
sédatif. Bien que Radix Astragali ait une longue histoire d'utilisation
médicinale en phytothérapie chinoise, ses
propriétés pharmacologiques et ses applications cliniques n'ont
été étudiées que récemment. Il a
été démontré que Radix Astragali a un large
éventail d'effets immunopotentiateurs et a été
utilisé comme médicament d'appoint pendant le traitement du
cancer. La demande de Radix Astragali est énorme à travers le
monde, en particulier sur le marché de l'Asie du Sud-Est et du Japon.
25
4.1.2 Glycyrrhizae Radix Et Rhizoma (Gancao)
Glycyrrhizae Radix Et Rhizoma (Gancao en chinois)
(Figure 21) est une parmi herbes les plus utilisé dans
la médecine traditionnelle chinoise en raison de ses divers effets
pharmacologiques et, plus important encore, des effets synergiques qui
améliorent l'efficacité et réduisent la toxicité
des autres herbes utilisés dans la médecine traditionnelle
chinoise (Shi, 2015). Les recherches qui sont été faites dans les
années passées ont montré que les effets
pharmacocinétiques de Gancao sont le résultat de ses constituants
tels que les macromolécules, comme les protéines, et les petites
molécules, comme les saponines et les flavonoïdes.

Figure 21. Illustration de la plante «
Glycyrrhizae Radix Et Rhizoma »
4.1.3 Radix saposhnikoviae (Fangfeng)
Cette herbe est utilisée dans la médecine
traditionnelle chinoise pour renforcer les défenses de l'organisme
contre les changements saisonniers, les polluants, les agents pathogènes
et les germes dispersés dans notre environnement (Figure
22) (Li, 2006). Elle est considérée l'une des herbes
traditionnelles les plus prisées à travers le paysage de la
médecine traditionnelle chinoise. Cette herbe aussi peut stimuler
l'immunité qui fonctionne de la saison de la grippe à la saison
des pollens, des avions sales aux collègues qui toussent, c'est l'herbe
qui combat l'immunité.

Figure 22. Illustration de la plante « Radix
saposhnikoviae »
26
4.1.4 Rhizoma Atractylodis Macrocephalae (Baizhu)
Cette herbe est un membre de la famille des tournesols qui se
développent naturellement dans les provinces du sud-est de la Chine
(Figure 23) (Li, 2007). Pendant des milliers d'années,
cette plante est très convoitée par les praticiens de la
médecine traditionnelle chinoise, souvent appelés «la
première plante revigorante de l'énergie vitale, y compris le
soutien immunitaire et l'équilibre émotionnel, la racine du Bai
Zhu a acquis une réputation puissante au cours des siècles. Cette
racine est utilisée pour aider à renforcer l'immunité et
à calmer les nerfs et les émotions.

Figure 23. Illustration de la plante « Rhizoma
Atractylodis Macrocephalae »
4.1.5 Forsythiae Fructus (Lianqiao)
Les recherches ont montré que les glycosides
phényléthanoïdes sont les principaux constituants bioactifs
de la Forsythiae Fructus (Figure 24). Ces constituants
bioactifs ont montré leurs effets anti-inflammatoires, antioxydants,
antibactériens et antiviraux . Forsythiae Fructus contient
jusqu'à trois cent et un de constituants. Tous ces constituants sont
listés avec leurs structures chimiques dans (Dong, 2017).

Figure 24. Illustration de la plante « Forsythiae
Fructus »
27
4.1.6 Lonicerae japonicae flos (Jinyinhua)
En médecine traditionnelle chinoise, la fleur de
chèvrefeuille a des propriétés douces et froides et est
associée aux méridiens du poumon, de l'estomac et du gros
intestin (Figure 25) (LI, 2015). Elle est utilisé pour
éliminer la chaleur et éliminer les toxines.
Généralement, la fleur de chèvrefeuille a
été utilisée pour traiter une variété de
conditions, allant de la fièvre, des ulcères, de l'inflammation
et des maux de gorge aux infections cutanées. Il est également
utilisé (en association avec les coptis et les pulsatilles) pour traiter
la diarrhée causée par la chaleur toxique. La fleur de
chèvrefeuille peut être appliquée en interne ou en
externe.
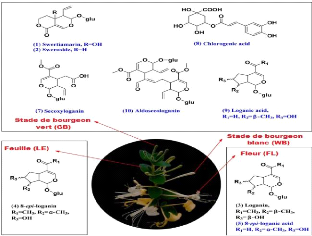
Figure 25. Illustration du chèvrefeuille et sa
structure moléculaire
4.2 Les médicaments anti-coronavirus à base
de la médecine traditionnelle Marocaine
La médecine traditionnelle marocaine est très
ancienne. Elle est riche par son expérience basée sur une culture
populaire dont elle est la manifestation concrète sous forme d'un
ensemble de pratiques relatives aux soins, à l'hygiène, à
la prévention et d'une façon générale à la
lutte contre la maladie. Elle s'adresse aux différentes pathologies en
utilisant par les pratiques traditionnelles accompagnées essentiellement
par une utilisation des plantes médicinales marocaines. Récemment
des efforts considérables sous forme d'enquêtes informatiques ont
été faits pour montrer la possibilité des
médicaments traditionnels marocains naturels extraits des herbes, de
lutter contre le virus "SARS-CoV-2" (Aanouz, 2020). Ces efforts avaient un
objectif précis visant à faire une sélection des plantes
étudiées selon deux principes: le premier est l'efficacité
orale, cela signifie que la majorité des plantes marocaines doivent
être absorbables par voie orale, le second principe est la
compatibilité des usages traditionnels, ensuite une utilisation
l'amarrage moléculaire a été faite pour avoir des
composés ayant un effet anti-coronavirus en se basant sur plusieurs
critères, par exemple l'énergie d'interaction. Les
résultats de l'amarrage moléculaire ont montré que parmi
67 molécules d'origine naturelle, trois molécules (Crocine,
Digitoxigénine et â-Eudesmol) sont proposées comme
inhibiteurs contre le coronavirus en se basant sur l'énergie
d'interaction
28
entre ces molécules et la protéine
étudiée « la protéine spike ». Les
résultats trouvés pour chaque molécule d'origine naturelle
en termes d'énergie d'interaction sont listés comme suit:
Tableau 1: L'interaction la plus importante de la liaison
hydrogène entre les trois composés naturels et la protéine
de pointe du coronavirus (2019-nCoV).
|
Type d'interaction
|
Liaison hydrogène pour Autodock vina
tool
|
|
â-Eudesmol
|
THR 111
|
|
Digitoxigenin
|
GLN 110, ASP 135
|
|
Crocin
|
THR 135, ASN 133, THR 199, LYS 137, LYS 5, PHE 3, ARG 4,
ARG 131
|
4.2.1 Digitoxigenin
La Digitoxigénine représente 11,25% de la
quantité présente dans le Nerium Oleander, les
dérivés de ces molécules sont utilisés comme
inhibiteurs antiviraux et anti-cancéreux (Boff, 2019). La structure
moléculaire de ce composé est représentée comme
suit (Figure 20 et Figure 26).
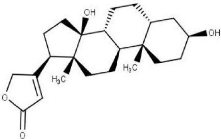
Figure 26. Illustration de la structure
moléculaire de digitoxigenin.
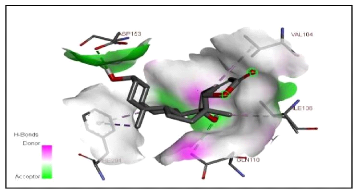
Figure 27. Illustration de la structure
moléculaire de Digitoxigenin en 3d.
29
4.2.2 f-Eudesmol
Le â-Eudesmol malgré sa faible quantité
dans la plante Lauris Nobilis L qui ne contient que 2,39%, ce composé
possède des pouvoirs antibactériens et antiviraux importants
(ASTANI, 2011). Sa structure moléculaire est représentée
comme suit (Figure 28 et Figure 29).
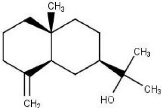
Figure 28. Illustration de la
structure moléculaire de f-Eudesmol.
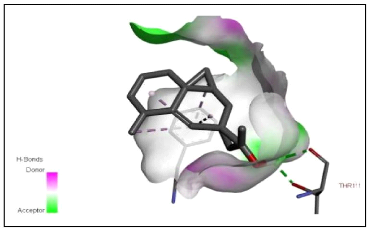
Figure 29. Illustration de la structure
moléculaire de f-Eudesmol en 3d.
4.2.3 Crocin
Le Crocin est un composé important dans le crocus
sativus L, il a la capacité d'inhiber la réplication du HSV avant
et après l'entrée des virions dans les cellules Vero. La crocine
pourrait être un agent anti-HSV et anti-VIH prometteur pour la
phytothérapie contre les infections virales (Soleymani, 2018). Sa
structure moléculaire est représentée comme suit
(Figure 31 et Figure 32).
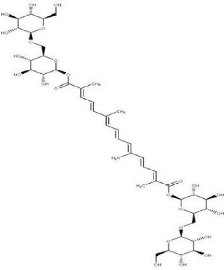
30
Figure 30. Illustration de la structure
moléculaire de Crocin.
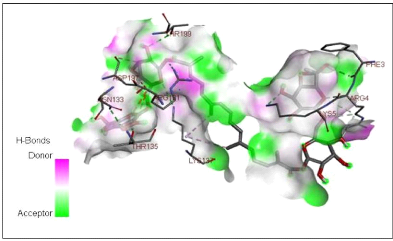
Figure 31. Illustration de la structure
moléculaire de Crocin en 3d.
31
5. Conclusion
Dans ce chapitre, nous avons introduit une révision
d'approches adoptées dans la recherche de nouveaux médicaments de
"SARS-CoV-2". Ensuite, nous avons s'intéressé à
l'identification de médicaments antibiotiques de "SARS-CoV-2" due a son
importance en l'absence d'une thérapeutique antivirale et d'un vaccin
spécifiques. Finalement, nous avons mis un éclairage sur les
médicaments anti-coronavirus à base de la médecine
traditionnelle chinoise et Marocaine.
32
Conclusion générale
Nous avons introduit une révision fondamentale et
extensive de plusieurs approches adoptées dans la recherche de nouveaux
médicaments. Cette révision détaillé nous a permet
clairement de faire une comparaison entre les différentes approches
existantes et leur efficacité de réponse aux cas d'urgence comme
celle lié avec virus "SARS-CoV-2". Le challenge pour ces approches et
pour les chercheurs c'est comment identifier un médicament efficace pour
lutter contre le virus "SARS-CoV-2" connu par sa propagation très.
Notre objectif principal était d'identifier les
approches qui ont surement la capacité de donner de bons
résultats surtout en termes du temps comme la propagation de «
SARS-CoV-2 » présente un danger très élevé due
à sa vitesse de propagation dans le monde entier. Dans le premier
chapitre, nous avons considéré premièrement la
modélisation avec les méthodes QSAR/QSPR due à la leur
capacité d'être largement utilisables dans le contexte de
recherche de nouveaux médicaments en fournissant une large gamme de
descripteurs qui peuvent servir des objectifs complémentaires entre
différents approches comme par exemple l'adoption de méthodes
QSAR/QSPR dans une approche basée sur la technologie in silico (Di
Tullio, 2012). Dans le deuxième chapitre, nous avons introduit plusieurs
approches à fin de mettre en éclairage leurs démarches
utilisés pour chercher de nouveaux médicaments et aussi leur
capacité à répondre aux besoins le plus tôt
possible. En plus, nous avons introduit la structure moléculaire des qui
font partie de chaque médicament identifié.
Nos efforts nous a permet de conclure que les approches
basés sur la médecine traditionnelle avaient
démontré de l'intérêt énorme dans ce contexte
et une réponse très rapide aux besoins. Plusieurs recherches ont
montré que la médecine traditionnelle est capable de lutter
contre le virus "SARS-CoV-2" et prévenir sa propagation. Dans ce
contexte nous avons met en éclairage surtout la médecine
traditionnelle chinoise via une exposition de plusieurs médicaments
identifiés capable être lutter contre le virus "SARS-CoV-2".
Nous efforts aussi nous conduit à identifier que la
médecine traditionnelle Marocaine à réussi à donner
de bons résultats. Grace à une étude expérimentale
est basée d'éléments d'origine naturelle extraits des
herbes. En faisant l'amarrage moléculaire de 67 molécules, donc,
trois molécules (Crocine, Digitoxigénine et â-Eudesmol)
sont identifiées comme inhibiteurs contre le coronavirus en se basant
sur l'énergie d'interaction entre ces molécules et la
protéine étudiée « la protéine spike
».
33
Bibliographie
Aanouz I., Belhassan, A., El Khatabi, K., Lakhlifi, T.,
El Idrissi, M., & Bouachrine, M Moroccan Medicinal plants as
inhibitors of COVID-19: Computational investigations [Journal]. - [s.l.] :
Journal of Biomolecular Structure and Dynamics, 2020. - 1-12 : Vols.
(just-accepted).
ASTANI Akram, REICHLING, Jürgen, et SCHNITZLER, Paul
Screening for antiviral activities of isolated compounds from
essential oils [Journal]. - [s.l.] : Evidence-based complementary and
alternative medicine, 2011. - Vol. vol. 2011.
Balaban A.T. Highly discriminating
distance-based topological index [Journal] // Chemical Physics Letters. -
[s.l.] : Chemical Physics Letters, 1982. - 399-404 : Vol. 89(5). - pp. 89,
399-404.
Barreiro E. J Privileged Scaffolds in Medicinal
Chemistry [Journal] // Royal Society of Chemistry. - London, 2015.
Bauer J., Fontain, E., & Ugi, I
Computer-assisted bilateral solution of chemical problems and
generation of reaction networks [Journal] // Analytica chimica acta. - [s.l.] :
Analytica chimica acta, 1988. - 123-134 : Vol. 210. - pp. 210, 123-134..
Bemis G. W. and Murcko M. A The properties of
known drugs. 1. Molecular frameworks [Journal] // J Med Chem. - [s.l.] :
Journal of medicinal chemistry, 1996. - 2887-2893 : Vol. 39(15). - pp. 39,
2887-93..
Boff L., Munkert, J., Ottoni, F. M., Schneider, N. F. Z.,
Ramos, G. S., Kreis, W., ... & de Padua, R. M Potential
anti-herpes and cytotoxic action of novel semisynthetic
digitoxigenin-derivatives [Journal]. - [s.l.] : European journal of medicinal
chemistry, 2019. - 54 : Vol. 167.
Breneman C.M., Sundling, C.M., Sukumar, N., Shen, L.,
Katt, W.P., and Embrechts, M.J New developments in PEST shape/property
hybrid descriptors [Journal]. - [s.l.] : Journal of Computer Aided Molecular
Design, 2003. - 231-240 : Vol. 17.
C.A. Lipinski F. Lombardo, B.W. Dominy, and P.J. Feeneyl
Experimental and computational approaches to estimate solubility and
permeability in drug discovery and development settings [Journal] // Advanced
Drug Delivery Reviews. - [s.l.] : Advanced drug delivery reviews, 1997. - 3-25
: Vols. 23(1-3). - pp. 6(1-3), 3-25.
CHEN Calvin Yu-Chian TCM Database@ Taiwan: the
world's largest traditional Chinese medicine database for drug screening in
silico [Journal]. - [s.l.] : PloS one, 2011. - no 1 : Vol. vol. 6.
Clark T QSAR and QSPR based solely on surface
properties? [Journal] // Journal of Molecular Graphics & Modelling. - 2004.
- pp. 22, 519-525.
34
CLYTI E., COUPPIE, P., STROBEL, M., et al
Traitement court de la donovanose par l'azithromycine. In : Annales de
dermatologie et de vénéréologie [Journal]. - [s.l.] :
Elsevier Masson, 2004. - p. 461-464 : Vol. No. 5.
CONSONNI Viviana, TODESCHINI, Roberto, PAVAN, Manuela, et
al Structure/response correlations and similarity/diversity analysis
by GETAWAY descriptors. 2. Application of the novel 3D molecular descriptors to
QSAR/QSPR studies [Journal]. - [s.l.] : Journal of chemical information and
computer sciences, 2002. - 693-705 : Vol. 42(3).
Cvetkovic R. S., & Goa, K. L
Lopinavir/ritonavir [Journal]. - [s.l.] : Drugs, 2003. - 769-802 :
Vol. 63(8).
D. Jaiswal C. Karthikeyan, and P. Trivedi
Rationalization of physicochemical properties of alkanoic acid
derivaties towards histone deacetylase inhibition [Journal] // Internet
Electronic Journal of Molecular Design. - [s.l.] : Internet Electron. J. Mol,
2006. - 13-26 : Vols. Des, 5. - pp. 5, 13-26.
de Groot R. J., Baker, S. C., Baric, R. S., Brown, C. S.,
Drosten, C., Enjuanes, L., ... & Perlman, S Commentary: Middle
East respiratory syndrome coronavirus (MERS-CoV): announcement of the
Coronavirus Study Group [Journal]. - [s.l.] : Journal of virology , 2013. -
7790-7792 : Vol. 87(14).
Dearden J.C In silico prediction of ADMET
properties: How far have we come? [Journal] // Expert Opinionon Drug Metabolism
& Toxicology. - 2007. - pp. 3 (5), 635-639.
Denis Fourches Modèles multiples en
QSAR/QSPR : Développement de nouvelles approches et leurs applications
au design "in silico" de nouveaux extractants de métaux, aux
propriétés ADMETox ainsi qu'à différentes
activités biologiques de molécules organiques [Report]. -
Strasbourg : l'Université Louis Pasteur (Strasbourg), 2007.
Di Tullio M., Maccallini, C., Ammazzalorso, A.,
Giampietro, L., Amoroso, R., De Filippis, B., ... & Kaliszan, R
QSAR, QSPR and QSRR in terms of 3-D-MoRSE descriptors for in silico
screening of clofibric acid analogues [Journal]. - [s.l.] : Molecular
informatics, 2012. - 453-458 : Vols. 31(6-7).
Dong Z., Lu, X., Tong, X., Dong, Y., Tang, L., & Liu,
M Forsythiae fructus: A review on its phytochemistry, quality control,
pharmacology and pharmacokinetics [Journal]. - [s.l.] : Molecules, 2017. - 1466
: Vol. 22(9).
Dudek Arkadiusz Z., Tomasz Arodz, and Jorge Gálvez
Computational methods in developing quantitative structure-activity
relationships (QSAR): a review [Journal]. - [s.l.] : Combinatorial chemistry
& high throughput screening, 2006. - 213-228 : Vol. 9(3).
Duval J., & Soussy, C. J. (1973)
Activité antibactérienne et pharmacocinétique de
l'amoxicilline. Comparaison avec l'ampicilline [Journal]. - [s.l.] :
Médecine et Maladies Infectieuses, 1973. - 525-531 : Vol. 3(12).
Euler L. the solution of a problem relating to
the geometry of position [Journal] // Commentarii academiae scientiarum
Petropolitanae. - [s.l.] : Commentarii Academiae Scientiarum Imperialis
Petropolitanae, 1976. - 123 : Vols. 8(128-140). - pp. 8, 128-140.
35
Fernández M. and Caballero, J QSAR models
for predicting the activity of non-peptide luteinizing hormone-releasing
hormone (LHRH) antagonists derived from erythromycin A using quantum chemical
properties [Journal]. - [s.l.] : Journal of Molecular Modeling, 2007. - 465-476
: Vol. 13.
Furuta Y., Gowen, B. B., Takahashi, K., Shiraki, K.,
Smee, D. F., & Barnard, D. L Favipiravir (T-705), a novel viral
RNA polymerase inhibitor [Journal]. - [s.l.] : Antiviral research, 2013. -
446-454 : Vol. 100(2).
Gilbert B. E., & Knight, V. E. R. N. O. N
Biochemistry and clinical applications of ribavirin [Journal]. -
[s.l.] : Antimicrobial agents and chemotherapy, 1986. - 201 : Vol. 30(2).
Golbraikh A., & Tropsha, A Beware of q2!
[Journal]. - [s.l.] : Journal of molecular graphics and modelling, 2002. -
269-276 : Vol. 20(4).
GUAN Wei-jie, NI, Zheng-yi, HU, Yu, et al
Clinical characteristics of 2019 novel coronavirus infection in China
[Journal]. - [s.l.] : MedRxiv, 2020.
Hasegawa K. and Funatsu, K Advanced PLS
techniques in chemoinformatics studies [Journal]. - [s.l.] : Current
computer-aided drug design, 2010. - 103-127 : Vol. 6(2).
HUANG Chaolin, WANG, Yeming, LI, Xingwang, et al
Clinical features of patients infected with 2019 novel coronavirus in
Wuhan [Journal]. - China : The lancet, 2020. - p. 497-506 : Vols. vol. 395, no
10223.
Isaacs A Interferon [Journal]. - [s.l.] : In
Advances in virus research, Academic Press, 1964. - pp. 1-38 : Vol. Vol. 10.
Karelson Mati Molecular descriptors in QSAR/QSPR
[Journal]. - New York : Wiley-Interscience, 2002. - Vol. 230.
Khadidja BELLIFA Etude des relations
quantitatives structure-toxicité des composés chimiques à
l'aide des descripteurs moléculaires. « Modélisation
QSAR» [Report]. - [s.l.] : Doctorat Classique en chimie, 2015.
Kier L. B., Hall, L. H., Murray, W. J., & Randi, M
Molecular connectivity I: Relationship to nonspecific local anesthesia
[Journal]. - [s.l.] : Journal of pharmaceutical sciences, 1975. - 1971-1974 :
Vol. 64(12).
Kumar S Drug and vaccine design against Novel
Coronavirus (2019-nCoV) spike protein through Computational approach [Journal].
- 2020.
L. He P.C. Jurs Assessing the reliability of a
QSAR model's predictions [Journal]. - [s.l.] : Journal of Molecular Graphics
and Modelling, 2005. - 503-523 : Vol. 23(6).
L. Zhang H. Zhu, T.I. Oprea, A. Golbraikh, A. Tropsha
QSAR Modeling of the Blood-Brain Barrier Permeability for Diverse
Organic Compounds [Journal]. - [s.l.] : Pharmaceutical research, 2008. -
1902-1914 : Vol. 25.
Leroy E. M., Kumulungui, B., Pourrut, X., Rouquet, P.,
Hassanin, A., Yaba, P., ... & Swanepoel, R Fruit bats as
reservoirs of Ebola virus [Journal]. - [s.l.] : Nature, 2005. - 575576 : Vol.
438(7068).
36
Li W., Li, L., LIU, Y. Y., YIN, X. J., & XIAO, Y. Q
Experimental study of pharmacological action of active composition of
Radix Saposhnikoviae [Journal]. - [s.l.] : Chinese Journal of Experimental
Traditional Medical Formulae, 2006. - Vol. vol. 6.
Li W., Wen, H. M., Cui, X. B., Zhang, F. C., & Dong,
J Chemical constituents in rhizome of Atractylodes macrocephala
[Journal]. - [s.l.] : Chinese Traditional and Herbal Drugs, 2007. - 1460-1462 :
Vol. 38(10).
LI Yujie, CAI, Weiyan, WENG, Xiaogang, et al
Lonicerae Japonicae Flos and Lonicerae Flos: a systematic pharmacology
review [Journal]. - [s.l.] : Evidence-Based Complementary and Alternative
Medicine, 2015. - Vol. vol. 2015.
Liu R., Sun, H., and So, S.-S Development of
quantitative structure-property relationship models for early ADME evaluation
in drug discovery: Part 2. Blood-brain barrier penetration [Journal] // Journal
of Chemical Information and Computer Sciences. - 2001. - pp. 41 (6),
1623-1632.
Ma X. Q., Shi, Q., Duan, J. A., Dong, T. T., & Tsim,
K. W Chemical analysis of Radix Astragali (Huangqi) in China: a
comparison with its adulterants and seasonal variations [Journal]. - [s.l.] :
Journal of agricultural and food chemistry, 2002. - 4861-4866 : Vol. 50(17).
Mak K. K., & Pichika, M. R Artificial
intelligence in drug development: present status and future prospects
[Journal]. - [s.l.] : Drug discovery today, 2019. - 773-780 : Vol. 24(3).
McClellan K., & Perry, C. M Oseltamivir
[Journal]. - [s.l.] : Drugs, 2001. - 263-283 : Vol. 61(2).
Mezey P.G Shape in Chemistry [Journal] //
Wiley-VCH. - 1993, New York.
Mezey P.G Shape in chemistry: an introduction to
molecular shape and topology [Journal] // Wiley-VCH, New York. - New York :
Wiley-VCH, 1993.
Mezey P.G The holographic electron density
theorem and quantum similarity measures [Journal]. - [s.l.] : Molecular
Physics, 1999. - 169-178 : Vol. 96 (2).
Mezey P.G The holographic electron density
theorem and quantum similarity measures [Journal] // Molecular Physics. - 1999.
- pp. 96 (2), 169-178.
Nakagawa T., Miyao T. and Funatsu K
Identification of Bioactive Scaffolds Based on QSAR Models [Journal]
// Mol Inform. - [s.l.] : Molecular informatics, 2018. - 1700103 : Vols.
37(1-2). - p. 37.
Organization World Health WHO Director-General's
remarks at the media briefing on 2019-nCoV on 11 February 2020. [Online] //
https://www.who.int/dg/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-20.
Qu X. A., & Rajpal, D. K. (2012)
Applications of Connectivity Map in drug discovery and development
[Journal]. - [s.l.] : Drug discovery today, 2012. - 1289-1298 : Vols.
17(23-24).
Rekkab Seifeddine Drug Design et synthèse
de nouveaux calixarènes sulfoniques flexibles à activités
anticorrosive et anticoagulante [Report]. - Alger : Thèse de Doctorat,
Univ. Constantine, 2014.
37
ROY Kunal, KAR, Supratik, et DAS, Rudra Narayan
Statistical methods in QSAR/QSPR [Book]. - [s.l.] : Springer, Cham,
2015. - Vols. (pp. 37-59) : pp. p. 37-59..
Samir Chtita Modélisation de
molécules organiques hétérocycliques biologiquement
actives par des méthodes QSAR/QSPR. Recherche de nouveaux
médicaments [Report]. - [s.l.] : (Doctoral dissertation), 2007.
Schultz H.P. Topoligical organic chemistry:
Graph theory and topological indices of alkanes [Journal] // Journal of
Chemical Information and Modeling. - [s.l.] : Journal of Chemical Information
and Computer Sciences, 1989. - 227-228 : Vol. 29(3). - pp. 29, 227-228.
Scott D. E., Coyne, A. G., Hudson, S. A., & Abell, C
Fragment-based approaches in drug discovery and chemical biology
[Journal]. - [s.l.] : Biochemistry, 2012. - 4990-5003 : Vol. 51(25).
Shi L., Tang, X., Dang, X., Wang, Q., Wang, X., He, P.,
... & Zhang, Y Investigating herb- herb interactions: The
potential attenuated toxicity mechanism of the combined use of Glycyrrhizae
radix et rhizoma (Gancao) and Sophorae flavescentis radix et rhizoma (Gancao)
and Sophorae flavescentis radix (Kushen) [Journal]. - [s.l.] : Journal of
Ethnopharmacology, 2015. - 243-250 : Vol. 165.
SMITH Amy, PENNEFATHER, Philippa M., KAYE, Stephen B., et
al Fluoroquinolones [Journal]. - [s.l.] : Drugs, 2001. - p. 747-761 :
Vols. vol. 61, no 6.
Soleymani S., Zabihollahi, R., Shahbazi, S., &
Bolhassani, A Antiviral effects of saffron and its major ingredients
[Journal]. - [s.l.] : Current drug delivery, 2018. - 698-704 : Vol. 15(5).
STRUPP Michael, ZINGLER, Vera Carina, ARBUSOW, Viktor, et
al Methylprednisolone, valacyclovir, or the combination for vestibular
neuritis [Journal]. - [s.l.] : New England Journal of Medicine, 2004. - p.
354-361 : Vols. vol. 351, no 4.
Tropsha A., Gramatica, P., & Gombar, V. K
The importance of being earnest: validation is the absolute essential
for successful application and interpretation of QSPR models
[Journal]. - [s.l.] : QSAR & Combinatorial Science, 2003. -
69-77 : Vol. 22(1).
Van Doremalen N., Bushmaker, T., Morris, D. H., Holbrook,
M. G., Gamble, A., Williamson, B. N., ... & Lloyd-Smith, J. O
Aerosol and surface stability of SARS-CoV-2 as compared with
SARS-CoV-1 [Journal]. - [s.l.] : New England Journal of Medicine, 2020. -
1564-1567 : Vol. 382(16).
Varin T. [et al.] Mining for bioactive scaffolds
with scaffold networks: improved compound set enrichment from primary screening
data [Journal] // J Chem Inf Model. - [s.l.] : Journal of chemical information
and modeling, 2011. - 1528-1538 : Vol. 51(7). - pp. 51, 1528-38.
Wang M., Cao, R., Zhang, L., Yang, X., Liu, J., Xu, M.,
... & Xiao, G Remdesivir and chloroquine effectively inhibit the
recently emerged novel coronavirus (2019-nCoV) in vitro [Journal]. - [s.l.] :
Cell research, 2020. - 269-271 : Vol. 30(3).
WHO World Health Organization Situation
Report-29 [Online] // Situation Report-29. - 2020. -
https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200218-sitrep-29-covid-19.pdf?sfvrsn=6262de9e_2.
38
Wiener H. Structural determination of paraffin
boiling points [Journal] // Journal of Chemical Information and Modeling. -
[s.l.] : Journal of the American Chemical Society, 1947. - 17-20 : Vol. 69(1).
- pp. 69, 17-20..
Yang Y., Islam, M. S., Wang, J., Li, Y., & Chen, X
Traditional Chinese medicine in the treatment of patients infected
with 2019-new coronavirus (SARS-CoV-2): a review and perspective [Journal]. -
[s.l.] : International journal of biological sciences, 2020. - 1708 : Vol.
16(10).
Yap C.W. and Chen, Y.Z Prediction of cytochrome
P450 3A4, 2D6, and 2C9 inhibitors and substrates by using support vector
machines [Journal] // Chemical Information and
Modeling. - 2005. - pp. 45, 982-992.
ZHAVORONKOV Alex, ALADINSKIY, Vladimir, ZHEBRAK,
Alexander, et al Potential COVID-2019 3c-like protease inhibitors
designed using generative deep learning approaches [Journal]. - [s.l.] :
Insilico Medicine Hong Kong Ltd A, 2020. - p. E1 : Vol. vol. 307.


