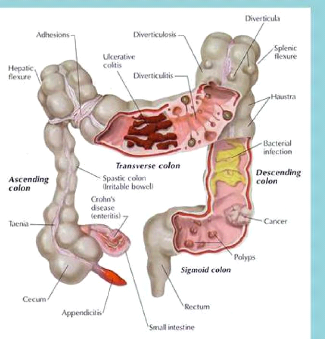
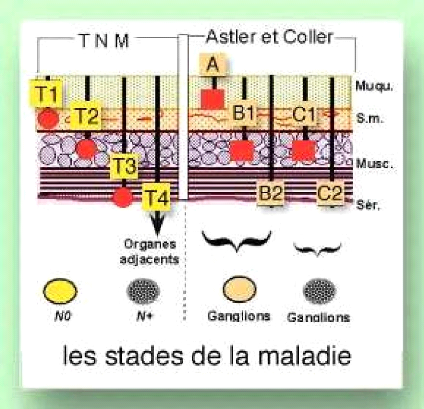
Apport du dosage de l'ACE dans le dépistage et la prévention du cancer du colon( Télécharger le fichier original )par Fatima Benhamza Université d'Oran es-senia - DES 2009 |

o Valeur pronostique en fonction du caractère sécrétant de la tumeur primitive Il est très rare que des cancers à concentration initiale d'ACE élevée rechutent sans augmentation du marqueur. La sensibilité du test est supérieure si la tumeur primitive sécrète l'ACE, mais il existe des tumeurs non sécrétantes initialement dont la rechute sera marquée par des valeurs sériques d'ACE supérieurs à la valeur de référence. Dans une étude, au seuil de 5tg/l, la sensibilité de l'ACE pour le détection des rechutes qui est de 97% ( spécificité 88%) en cas d'ACE préopératoire supérieur à la valeur de référence, n'est plus que de 66% ( spécificité 94%) en cas d'ACE préopératoire inferieur à la valeur de référence. (Wang, 1994). Dans deux études menées chez des patients ayant une tumeur de stade C de Dukes initialement non sécrétant, la sensibilité d'une valeur sérique de l'ACE supérieure à la valeur de référence pour la détection des récidives est estimée à 36% (spécificité 93%) et 44%. Chez ces patients, l'augmentation de la concentration sérique du marqueur est pratiquement toujours associée à l'apparition de métastases viscérales. (Zeng, 1993) ; (Tobaruela, 1997). Pour certains auteurs, les tumeurs peu différenciées peuvent être suivies par le dosage de l'ACE si le marqueur est détecté par immunohistochimie dans la tumeur primitive. (Goslin, 1981) ; (Zeng, 1993). La valeur diagnostique de l'Ace varie en fonction du caractère sécrétant ou non de la tumeur mais un taux initial normal ne doit jamais exclure les marqueurs des paramètres de surveillance. o Valeur pronostique en fonction du site de récidive La sensibilité de l'ACE est d'environ 78% à 97% pour la détection des récidives intra hépatiques , de 75% pour les récidives rétro péritonéales, de 45% pour celles qui sont locorégionales et de 42 à 50% pour les métastases pulmonaires (Moertel, 1993) ; (Bergamashi, 1996).La sensibilité de l'ACE diminue en cas de métastases solitaires, dans l'étude de Moertel, elle est de 76% pour les métastases hépatiques solitaires alors que pour les métastases pulmonaires solitaires elle n'est que de 15%. La valeur diagnostic de l'ACE varie en fonction du site de la récidive. La sensibilité de l'ACE pour la détection des récidives est meilleure lorsque les récidives sont hépatiques que lorsqu'elles atteignent d'autres sites. o Valeur pronostique en fonction de la méthode d'appréciation des augmentations de l'ACE Plusieurs méthodes d'appréciation des augmentations des concentrations sériques de l'ACE ont été décrites. Elles peuvent être réparties en deux groupes, groupe des méthodes comparant le résultat à une valeur seuil et groupe utilisant la cinétique d'évolution des concentrations sériques de l'ACE. o Méthodes comparant le résultat à une valeur seuil Selon les pratiques, le seuil retenu pour la surveillance est un seuil individuel ou un seuil fixé pour l'ensemble des patients à partir de la valeur de référence (Tableau IV), (Lucha, 1997). Tableau IV : études ayant évalué la valeur diagnostique de l'ACE en fonction d'une méthode basée sur l'adoption d'une valeur seuil. (Lucha, 1997)
NB : nd= non défini L'interprétation des résultats de dosage de l'ACE en fonction d'un seuil individuel est le fait d'une équipe. Le seuil individuel est déterminé à partir de la ligne de base des concentrations de l'ACE post opératoire. Les augmentations de l'ACE sont interprétées avec un intervalle de confiance analytique de 95%. La valeur prédictive de cette méthode sur la découverte chirurgicale d'une récidive est de 88% dans la population initiale (Martin, 1977), mais le nombre important de faux positifs dus aux fluctuations individuelles en a limité la diffusion (Rittgers, 1978). La majorité des auteurs utilisent un seuil qui est égal à la valeur de référence ou à un multiple de cette valeur (Koch, 1982) ; (Lunde, 1982) ; (Szymendera, 1982) ; (Tate, 1982) ; (Carlson, 1983) ; (Graffner ; 1985) ; (Moertel, 1993) ; (Mc call, 1994) ; (Wang, 1994) ; (Lucha, 1997). o Lorsque le seuil est égal à la valeur de référence, la sensibilité pour la détection de récidive varie de 58% pour une spécificité de 93% à 90% pour une spécificité de 66%, la VPP varie de 41% à 79% et la VPN de 75% à 99%. o Lorsque le seuil est égal au double de la valeur de référence, la sensibilité pour la détection de récidive varie de 31% pour une spécificité de 96% à 78% pour une spécificité de 91%, la VPP varie de 65% à 89% et la VPN de 69% à 90%. L'analyse des études évaluant la fiabilité du test à plusieurs seuils d'interprétations montrent que l'adoption d'un seuil élevé permet d'améliorer la valeur prédictive du test, mais au prix d'une diminution importante de sa sensibilité (Steele, 1982) ; (Carlsson, 1983) ; (Denstman, 1986) ; (Moertel, 1993). o Méthodes utilisant une appréciation dynamique des concentrations sériques de l'ACE Plusieurs auteurs ont plaidé très tôt pour une appréciation dynamique des variations des concentrations sériques d'ACE, plus sensible que le seuil puisqu'elle permet de constater l'élévation du marqueur même dans la zone des valeurs normales, et plus spécifique puisqu'elle permet d'éliminer les variations transitoires des concentrations sériques du marqueur (Rittgers, 1978) ; (Staab, 1978). (Tableau V), (Mora, 1997). Tableau V : études ayant évalué la
valeur diagnostique de l'ACE en fonction
Pour certains auteurs, l'analyse cinétique se résume à la mise en évidence sur plusieurs prélèvements successifs d'une augmentation de la concentration de l'ACE par rapport à la ligne de base postopératoire (Szymendera, 1982) ; (Sugarbaker, 1987) ; (Mora, 1997) ; (Nakayama, 1997). Pour d'autres, l'analyse se pente intervient après transformation logarithmique log (1+ACE), ce qui met en évidence le caractère exceptionnel de l'augmentation du marqueur (Steele, 1982) ; (Boey, 1984) ; (Denstman, 1986), tab. Les malades qui demeurent en rémission clinique ont une pente d'évolution du marqueur sensiblement nulle. En cas de récidive, la valeur médiane de l'accroissement mensuel est de 5,8% dans l'étude de Steele et de 20% dans l'étude prospective de Boey. ( 20% de récidive en cas de récidive versus 0,3% en l'absence de récidive, p0,001). Une étude utilise un modèle mathématique tenant en compte de la croissance exponentielle de la tumeur, de la dilution du marqueur dans le compartiment vasculaire et de la contribution à la production du marqueur par le tissu normal. La sensibilité et la spécificité ne sont pas plus élevées que dans une approche cinétique plus simple (Carl, 1993). L'analyse de pente permet le calcul du temps de doublement du marqueur. Beaucoup d'autres s'accordent sur le fait que la perte d'accroissement est plus rapide en cas de métastase hépatique qu'en cas de récidive locale et que le temps de doublement pourrait constituer une aide pour la localisation des récidives ( Steele,1982) ; (Boey, 1984) ; (Carl,1993) ; (Umehara, 1993). Le temps de doublement du marqueur précédant le diagnostic de la rechute est un facteur pronostique sur la survie, une augmentation rapide des concentrations sérique du marqueur ayant une signification péjorative. (Staab, 1985) ; (Koreyema, 1997). Deux études comparant chez les mêmes patients l'efficacité de la comparaison, des résultats à différents seuils s et de l'analyse dynamique montrent que le meilleur accord entre la précision biologique de la récidive et la réalité est obtenue avec un seuil un peu supérieur à la valeur de référence mais que cet accord est inferieur à celui obtenu avec une analyse dynamique. (Steele, 1982) ;(Denstman, 1986). En conclusion : o L'ACE est le premier indicateur de récidive dans 65% des cas. o L'ACE est le marqueur de choix pour surveiller les patients atteints de cancer colorectal. o Il est très rare que des cancers à concentration sérique initiale d'ACE élevée rechutent sans augmentation du marqueur. o Il n'y a pas de méthodes standard pour apprécier l'augmentation des concentrations sériques d'ACE dans le cadre de la surveillance des patents après chirurgie curative. 1.6.4.3. Intérêt de la surveillance Une surveillance biologique soutenue incluant le dosage de l'ACE permet de prédire la survenue de récidive avec une avance de quelques semaines à quelques mois sur le diagnostique clinique ou radiologique, cette surveillance permet aussi le diagnostic de récidives à un stade où l'opérabilité est meilleure. Mais il n'existe pas à l'heure actuelle, à l'échelle d'une population de malades des suivis, de bénéfices démontrés, en temps de survie, résultant de cette avance au diagnostic et cette meilleure opérabilité. L'inclusion du dosage de l'ACE dans la surveillance des cancers du colon varie selon les comités d'experts : o Pas de surveillance se l'ACE en dehors d'essais thérapeutiques o Surveillance régulière de l'ACE chez les patients en état de supporter une thérapeutique autre que symptomatique. o En l'absence de consensus, la surveillance régulière des concentrations de l'ACE après chirurgie curative n'est pas recommandable systématiquement. La place du dosage de l'ACE dans le suivi des patients devra être évaluée à la publication études et au moment où des nouvelles techniques d'imagerie tel que le scanner hélicoïdal et de chirurgie tel le traitement percutané des métastases seront plus largement utilisées et que le traitement systématique du cancer aura gagné en efficacité. 2. Implication de la protéine â caténine-APC dans le développement du cancer du colon 2.1. Gène APC Localisé sur le bras long du chromosome 5, possédant 15 exons et codant pour une protéine de 311kda, constituée de 2844 acides aminés formant des homodiméres par sa partie aminoterminale. Elle est localisée au pole basolatéral de la cellule épithéliale colique. Son expression est d'autant plus importante que la cellule migre du fond de la crypte vers le sommet de la villosité. Outre la béta caténine, quatre partenaires de la protéine APC sont maintenant connus et forment avec elle des complexes protéiques. Il s'agit de la protéine EB1 de 30kda, dont la fonction est inconnue (Su et a!, 1995), la protéine DLG codée par un gène humain homologue du gène suppresseur de tumeur chez la drosophile « Disk large » (Matsumine et a!, 1996), de la glycogen synthase kinase 3béta (Rubinfield et al, 1996) et des microtubules composants du cytosquelette (Smithe et a!, 1994). Bien que l'interaction de la protéine APC avec EB1 et DLG soit probablement importante dans le processus de transformation maligne (puisque la plupart des altérations du gène APC dans les cellules tumorales coliques conduisent à la synthèse d'une protéine tronquée ne possédant pas ses domaines de liaisons), les avancées récentes sur la compréhension du rôle de la protéine APC sont venues de l'étude de la béta caténine. En effet, son interaction avec cette protéine semble être l'élément qui fait le lien entre deux processus apparemment différents ; l'adhésion et la prolifération cellulaire. (fig 12) (Rubinfield et a!, 1996).
Figure 12: structure du gène APC (Rubinfield et a!, 1996) 2.1.1. Mutations du gène APC et instabilité chromosomique L'analyse génétique des cancers colorectaux a permis de révéler l'existence de deux formes distinctes. La première retrouvée dans 15% des cancers, est associée à un défaut du système de réparation des mésappariements de l'ADN qui se traduit par une instabilité des microsatellites (Phénotypes MSI+ ou RER+) (Puig et al, 1999). La seconde, de loin la plus fréquente, puisque retrouvée dans 85% des cancers colorectaux, se caractérise par une instabilité chromosomique (Phénotype LOH+). L'origine de cette instabilité chromosomique n'était jusqu'à présent pas clairement identifié, mais deux articles récents montrent que les mutations de la protéine APC pourraient bien en être responsables. Deux équipes décrivent en effet dans Nature Cellul Biology que des cellules embryonnaires de souris homozygotes pour la mutation Min du gène APC acquièrent, au cours des passages en culture, des anomalies du nombre de chromosomes qui sont liées à un défaut de ségrégation des chromosomes. (Fodde et a!, 2001) ; (Kaplan et a!, 2001). L'allèle Apc min code pour une protéine tronquée de sa partie carboxyB terminale, qui ne peut alors ni se lier aux microtubules, ni participer à la dégradation de la béta caténine. D'autres mutations, comme la mutation Apc1638T, située dans une région plus distale, n'altèrent pas la fonction de dégradation de la béta caténine, mais provoquent aussi une instabilité chromosomique. Ceci suggère que cette instabilité n'est pas liée à un effet tanscriptionnel induit par le complexe béta caténine/TCF4. (Kaplan et a!, 2001). Les deux groupes observent que la protéine APC native est localisée, pendant la mitose, à l'extrémité positive des microtubules, au niveau des kinétochores. Il s'agit probablement d'un attachement physique de la protéine APC à l'extrémité des microtubules, car leur dépolarisation empêche cette localisation. La protéine mutée Apc min est incapable de se lier correctement aux kinétochores. Il existe alors une désorganisation du fuseau mitotique, de nombreux microtubules ne s'attachent plus aux kinétochores, et on observe parfois des centrosomes surnuméraires, ces anomalies étant très certainement à l'origine de l'instabilité chromosomique. L'interaction entre les microtubules et la protéine Apc se ferait par l'intermédiaire de la protéine EB1, dont la localisation normale au niveau des kinétochores disparaît dans les cellules exprimant la protéine Apc mutée. (Fodde et al. 2001). Les deux équipes émettent l'hypothèse d'un rôle de la protéine Apc dans la stabilisation des microtubules et leur attachement aux chromosomes. En outre, Apc interagit avec d'autres protéines du kinétochore, dont la protéine kinase BUB1qui intervient dans le point de contrôle mitotique. (Kaplan et al, 2001), cependant la signalisation de cette interaction n'est pas connue. La fonction de la protéine Apc n'est donc pas restreinte à celle de gène suppresseur de tumeur, mais pourrait aussi être essentielle à une ségrégation correcte des chromosomes lors de la mitose. Son altération pourrait aussi rendre compte de l'aneuploïdie observée lors de la transformation maligne des cellules. 2.1.2. Le gène APC, un partenaire essentiel de la voie Wnt La protéine Apc joue un rôle majeur dans le contrôle de l'activité de la voie de signalisation Wnt. Cette voie aboutit à la formation du complexe de transactivation béta caténine /TCF4, qui active la transcription de nombreux gènes cibles, en particulier l'oncogène C-Myc ou la cycline D1. La protéine Apc favorise la dégradation de la béta caténine au sein d'un complexe contenant aussi l'axine et la glycogène synthase kinase 3béta. La principale conséquence des mutations inactivatrices d'Apc est l'accumulation de la béta caténine dans le cytosol et le noyau et donc l'activation de la voie Wnt. S'il est bien établi qu'Apc a un rôle de gène suppresseur de tumeur, il semble cependant que ce ne soit pas sa seule fonction. En effet la protéine Apc peut se lier par sa partie carboxy-terminale, aux microtubules soit par l'intermédiaire de EB1, une protéine qui est elle-même associée à l'extrémité positive des microtubules, ceci suggérait que Apc puisse jouer un rôle dans l'organisation du cytosquelette ou dans celle du fuseau mitotique. (He et al. 1998). 2.2. Caractérisation d'une voie importante dans la signalisation de la carcinogénèse colorectale : voie Wnt 2.2.1. Signalisation Wnt/ â-caténine La voie de signalisation Wnt / wingless joue un rôle important au cours du développement embryonnaire en modulant l'expression des signaux intercellulaires qui contrôlent la croissance, la migration, le déterminisme et la polarisation cellulaire. (Bellaiche et perrimon, 1997). Elle a été particulièrement étudiée au cours du développement chez le nématode, la drosophile et les amphibiens, mais ses constituants sont très conservés chez les vertébrés. Dans les tissus adultes, elle participe au contrôle de la prolifération cellulaire. Une activation anormale de cette voie par mutation de l'un ou l'autre de ses composants joue également un rôle important dans un grand nombre de cancers humains. (Romagnolo, 1997). Cette voie est alors appelée souvent voie APC/béta caténine/TCF. Que ce soit au cours du développement ou de la carcinogenèse, la clé de voûte de cette voie est la béta caténine ; lorsque la voie est inactive, celle-ci est dégradée, lorsque la voie est active, elle est libérée dans le cytoplasme où elle exerce ses fonctions. (Romagnolo, 1997). Dans la position « off », la béta caténine est partie intégrante d'un complexe multiprotéique constitué de l'axine (ou son homologue le conductine), de la protéine phosphatase PP2A, de la glycogen synthase kinase 3béta et d'Apc. Au sein de ce complexe l'interaction entre l'axine et GSK3béta semble favoriser la phosphorylation de la béta caténine et sa dégradation ultérieure par le protéasome. La position « on » est obtenue de manière différente selon qu'il s'agit d'un processus normal du développement ou d'un processus de carcinogenèse. Dans le premier cas, l'activation des récepteurs Frizzled par la fixation de Wnt conduit à la phosphorylation de la protéine Dishevelled qui, par son association à l'axine, empêche la GSK3béta de phosphoryler ses substrats parmi lesquels se trouve le béta caténine, rendant ainsi la liberté à cette dernière. Dans les cancers, indépendamment de toute activation du récepteur Frizzled, c'est l'inactivation ou l'activation de gènes codants pour des protéines impliquées dans le complexe phosphorylant la béta caténine qui bloque la dégradation de béta caténine. Le plus souvent, il s'agit de mutation d'Apc mais il peut aussi s'agir de mutation au niveau de séquences répétées codantes du gène codant pour l'axine, ou celles activatrices, dans l'exons 3 du gène codant pour la béta caténine au niveau des sites de phosphorylation. Dans tous les cas, lorsqu'elle n'est pas dégradée, la béta caténine s'accumule dans le cytoplasme, une partie a un rôle dans l'adhérence cellulaire en s'associant avec les E-cadhérines et l'actine du cytosquelette pour former les fonctions gap. Une autre partie de la béta caténine libre est véhiculée dans le noyau où elle s'associe aux facteurs de transcription TCF/LEF, modulant ainsi leur activité et activant l'expression d'un grand nombre de gènes cibles (Fig13) , (Behrens et al, 1996) ; (Korinek et al, 1997).
Figure 13: représentation schématique des différents partenaires de la béta caténine (Korinek et al, 1997). 2.2.2. La â caténine l'élément clé dans les mécanismes de régulation de la voie Wnt Il y'a peu de temps encore, on limitait le rôle de la béta caténine et son homologue Armadillo, chez la drosophile, aux processus d'embryogenèse et aux mécanismes cellulaires permettant l'adhésion des cellules entre elles. (Peifer et al, 1997) ; (Resnik et al, 1997). En effet la béta caténine est d'une part, l'un des éléments du signal de transduction qui participe au développement axiale de l'embryon, et d'autre part, l'un des composants des jonctions adherens. (Heasman et al, 1994) ; (Kemler et al, 1993). Plus récemment l'étude de la cancérogenèse colique. En particulier, l'intérêt qui lui a été porté dans la carcinogenèse colique a permis de montrer que la béta caténine semble être un acteur majeur de la transformation maligne d'une cellule épithéliale colique. En particulier, l'intérêt qui lui a été porté dans la carcinogenèse colorectale a commencé à prendre de l'ampleur lorsqu'elle a été reconnue comme l'un des partenaires cellulaires de la protéine Apc. (Su et al, 1993) ; (Rubinfield et al, 1995). Les mutations du gène de la béta caténine affectent la région aminoterminale de la protéine la rendant réfractrice à sa régulation par APC. La béta caténine n'est pas essentiellement dans le colorectal mais aussi dans de nombreux autres cancers. (Piard et al, 2002). 2.2.2.1. Structure de la â caténine Trois domaines importants ont été identifiés sur la béta caténine o Le domaine N-terminale, composé de 149 acides aminés, possédant de nombreux sites de phosphorylation par GSK-3b, par la caséine Kinase (CKIa) et un site de fixation (sérine 33/37) à la protéine b-TrCP initiatrice du processus de dégradation par le protéasome. o Domaine central comprenant 12 ARD (Armadillo Repeat Domain) de 42 acides aminés chacun. Reconnaissant notamment la protéine Apc, l'axine/conductine ou encore les facteurs de transcription de la famille TCF. o Un domaine C-terminal de 108 acides aminés ayant une fonction, lui permettant de jouer au niveau nucléaire son rôle d'activateur de la transcription (Fig 14.) (Peifer, 1997).
Figure14: structure de la â caténine (Peifer, 1997). 2.2.2.2. Rôle de la â caténine Dans une cellule normale et en l'absence des facteurs de la famille wnt la concentration de béta caténine est maintenue à un niveau très faible par une dégradation constitutive de la protéine par le protéasome. (Aberle et a!, 1997). Dans les processus de développement, les facteurs de la famille wnt induisent, par l'intermédiaire de leurs récepteurs membranaires Frizzled et l'activation de la protéine cytoplasmique intermédiaire Dishevelled, une déstabilisation de ce complexe en inhibant l'activité de la GSK3béta. L'élévation du niveau de béta caténine libre cytoplasmique dans les cellules tumorales coliques est suivie de la translocation nucléaire de la protéine. La béta caténine ne possédant pas de séquence de nucléarisation, pourrait être transloquée vers le noyau en s'associant aux facteurs Tcf/Lef via son domaine ARD. (Behrens et al, 1996) ; (huber et a!, 1996). Cette translocation peut cependant être indépendante des facteurs Tcf/Lef. (Fogotto et a!, 1998). Dans le noyau, le complexe béta caténine/Tcf-Lef se lie à des gènes cibles par l'intermédiaire du domaine de liaison à l'ADN des facteurs Tcf/Lef, alors que les domaines d'activations transcriptionnelle sont apportés par la béta caténine. Dans les cellules coliques présentant des mutations d'APC ou de béta caténine, ce complexe active de façon constitutive la production de gènes dont les produits contribuent à la progression tumorale. (TableauVI) C-Myc est un facteur de transcription oncogénique dont les gènes cibles produisent des protéines impliquée, notamment dans le régulation du cycle cellulaire et de l'apoptose. Le gène C-Myc a récemment été identifié comme une cible directe du complexe de la béta caténine/Tcf-Lef. (He et a!, 1998). Son promoteur contient des séquences consensus de liaison à l'un des facteurs de la famille Tcf, « cf-4 binding élément ». Il est surexprimé dans les cellules tumorales coliques présentant une mutation d'APC ou de béta caténine. L'activation incontrôlée du complexe transcriptionel béta caténine/Tcf-Lef expliquerait donc la surexpression de C-Myc très fréquemment dans les formes héréditaires ou sporadiques des cancers colorectaux. La cycline D1, un autre régulateur important du cycle cellulaire, a été identifié comme une cible directe du complexe béta caténine_Tcf. (Shtutman et a!, 1999). Cela expliquerait sa surexpression dans 30% des adénocarcinomes et polypes adénomateux du colon. La matrilysine est une protéine de la famille des métalloprotéinases. Elle est impliquée dans les processus de dégradation de la matrice extracellulaire nécessaire à l'invasion tumorale et la formation e métastase. Deux sites de liaison à Tcf-4 ont été identifiés sur son promoteur et sa transcription est activée par le complexe béta caténine-Tcf-4 (Crawford et a!, 1999). La matrilysine est surexprimée dans 80% des adénomes et des carcinomes coliques humains résultant de la mutation générale ou somatique d'APC. Son expression est totalement corrélée à celle de la béta caténine dans les tumeurs. Elle est également retrouvée dans les polypes précancéreux, contrairement aux autres métalloprotéinases qui sont exprimées tardivement au cours de la carcinogenèse. La gastrine, puisant stimulant de la sécrétion acide gastrique, est également un facteur de croissance pour les cellules tumorales coliques. Les produits issus s'une mutation post-traductionnelle incomplète de l'hormone surexprimés dans les cancers du colon, jouent un rôle important dans la prolifération des cellules normales ou tumorales de la muqueuse colique. (Van Solinge et a!, 1993) : (Wang et a!, 1996) ; (Koh et a!, 1999). L'analyse du gène de la gastrine a révélée l'existence de plusieurs sites de liaisons potentiels du facteur Tcf. (Koh et a!, 2000). Sur des cellules en cultures, l'expression d'une forme constitutivement active de béta caténine augmente l'expression du gène de la gastrine. Inversement, la transfection d'un mutant dominant négatif TCF4 inverse cet effet. L'activation du gène de la gastrine par le complexe béta caténine-tcf4 pourrait donc contribuer à la progression tumorale des cellules coliques. Il semble claire que la béta caténine puisse être définie comme un oncogène jouant un rôle important dans la progression tumorale colique. Elle est constitutivement activée très précocement au cours de le carcinogenèse par les altérations géniques qui peuvent soit l'affecter directement, soit touché des protéines impliquées dans la régulation de sa dégradation tel que APC, GSK3béta. Son activation constitutive conduit à l'expression dérégulée des gènes cibles impliqués dans des processus cellulaires tel que la prolifération ou l'invasion qui ont un rôle important dans la progression tumorale (Fig 15) (Koh et a!, 2000).
Figure 15: role de la béta caténine (Koh et al, 2000). TableauVI : liste des gènes cibles de la â caténine dans les cancers du colon humain. (Kolligs et a!, 2002).
2.3. La régulation de l'activité de la â caténine 2.3.1. Les protéines GSK-3b et Axine Dans toutes les cellules normales, la sérine/thréonine GSK3b joue un rôle clé dans le processus de dégradation de la béta caténine. En phosphorylant la béta caténine sur plusieurs sites consensus situés dans son domaine aminoterminal, elle permet une reconnaissance de la protéine par des ubiquitines ligases. La béta caténine ubiquitinylée est alors rapidement dégradée par le protéasome. Plusieurs études ont notamment démontré que des délétions de la partie aminoterminale de la béta caténine conduisent à son accumulation dans le cytosol. In vitro, la béta caténine est peu phosphorylée par GSK3b car il n'y a pas d'interaction directe entre ces deux protéines. In vivo, dans le complexe protéique quaternaire : béta caténine, GSK3b et protéine Apc. Elle contribue au rapprochement de la ligase GSK3b et ses substrats et facilite leur phosphorylation. (Ikeda et a!, 1998) ; (Hart et a!, 1998). Dans ce complexe, la phosphorylation de la protéine Apc augmente son association avec la béta caténine et la phosphorylation de cette dernière. (Rubinfield et al, 1996). La phosphorylation de l'axine/conductine par GSK3b augmenterait la stabilité de la protéine. L'accumulation de la béta caténine dans les tumeurs colorectales résulte essentiellement d'altérations géniques de l'un de ses partenaires, mais cependant aucune mutation au niveau de GSK3b n'a été détectée (fig 16), (Reya, Clevers, 2005).
Figure 16 : Régulation de la â caténine la GSK-3b (Reya, Clevers, 2005). La GSK-3b est elle aussi sujette à de nombreux mécanismes de régulation. Elle peut être inhibée par phosphorylation en sérine9 (Tsujio et a!, 2000) ; (Grimes et Jope, 2001). De nombreuses kinases ont été décrites pour être responsable de ces modifications comme la p 70S6kinase, p 90rsk (leur effet peut être réversible par la PP2A) (Sutherland et a!, 1993), Akt (Cross et a!, 1995). ILK (Intergin Linked Kinase) (Delcommenne, 1998), des protéines kinases (Cook et a!, 1996) ; (Tsunjio et a!, 2000) et des protéines kinases dépendantes d'AMP cyclique (Protéine Kinase A) (Li et al, 2000). La formation de complexes protéiques, médiées par les protéines associées à la GSK-3b, est également un autre moyen pour la réguler. En effet, rappelons que l'association de la GSK-3b au sein du complexe APC/Axine/ â caténine est responsable de son activation. La fixation de Frat-1 sur la GSK-3b, facilitée par la protéine Disheveled, entraine son inhibition et sa séparation de l'axine (Li et a!, 1999) ; (Tsujo et a!, 2000) ; (Grimes et Jope, 2001). Enfin la GSK-3b peut être activée par phosphorylation en tyrosine 216, ce qui augmente son activité enzymatique. Le sort et son équipe ont montré que Fyn pourrait directement phosphoryler la GSK-3b en tyrosine 216 et donc l'activer. (Lesort et a!, 1999). De plus, une élévation transitoire de la concentration intracellulaire de Ca2+entraîne également l'activation de la GSK-3b (Hartigan et Johnson, 1999). 2.3.2. Régulation de la â caténine par APC L'Apc se lie à la béta caténine (Rubinfield et a!, 1993) ; (Su et a!, 1993) qui a été originellement associé à la zone d'adhérence des cellules épithéliales. La béta caténine se lie au domaine cytoplasmique des cadhérines et à l'alpha caténine. Elle est reliée au réseau d'actine via l'alpha caténine. L'Apc se lie à la béta caténine par deux régions différentes (fig 17) (Su et a!, 1993). Bien que de nombreux mutants de l'Apc aient perdu au moins l'un des deux sites de fixation, ils gardent la capacité de se lier à la béta caténine.
Figure 17: interaction entre béta caténine et Apc (Su et al, 1993). L'Apc pourrait jouer un rôle important dans l'adhérence cellulaire par ses capacités de liaison avec la béta et gama caténine (plakoglobine) en modulant l'expression de béta caténine libre et fixée dans la cellule. En effet, certaines lignées de cellule du colon qui expriment l'Apc mutée contient des taux importants de béta caténine. La transfection de ces cellules avec l'Apc sauvage ou avec des fragments contenant la partie centrale des 20 acides aminés réduit significativement la quantité totale de la béta caténine. (Munemitsu et a!, 1995). Une surexpression de l'apc sauvage induit une augmentation du catabolisme de la béta caténine. La région de l'Apc responsable de la régulation de la béta caténine est localisée dans la partie centrale de la molécule, qui est également le domaine phosphorylable par la GSK3b. (Rubinfield et a!, 1996). La forme phosphorylée de l'Apc aurait une affinité plus importante pour la béta caténine que la forme non phosphorylée. L'hypothèse émise est que l'Apc « pressentirait » l'excès de béta caténine intracellulaire. Elle s'associerait alors plus fortement à la GSK3b qui la phosphoryle et à la béta caténine pour en démarrer la dégradation protéolytique, via la voie ubiquitine-protéasome. (Aberle et al, 1997). 2.3.3. La fixation de Disheveled au sein du complexe multi protéique Les analyses génétiques de Dsh l'ont défini comme un médiateur positif de la voie Wnt, positionné en aval du récepteur et en amont de la â caténine. (Noordemer et al, 1994). La famille de protéine Dsh ne possède pas de fonction enzymatique connue, mais comporte de nombreux sites d'interaction potentiels avec de nombreuses protéines ; un domaine NT DEP. Il a été décrit chez la drosophile, que l'expression ectopique de Dsh activait la voie Wnt (Yanagawa et al, 1995). Disheveled, a trouvé son homologue chez les mammifères (Dv1), ainsi que sa place au sein de la voie Wnt. En effet, l'interaction de la protéine Wnt avec Frizzled va activer la protéine Disheveled selon des mécanismes encore mal connus. En revanche, il a été montré après activation de la voie Wnt que DvI et l'axine interagissent par leurs domaines DIx respectifs (Kishida et al, 1999). La liaison Dv1 sur l'axine déstructure le complexe de dégradation. Les kinases n'ont donc plus accès à la â caténine qui peut ainsi s'accumuler dans le cytoplasme. (Sub et al, 2001). De plus, il a été démontré que la caséine kinase Ie active ( CKIe) régule positivement la voie de signalisation Wnt en déstabilisant le complexe de dégradation. Elle agit en aval de Dv1 mais en amont de la GSK-3b. (Gao et al, 2002). Une 3eme kinase, Par-1, est également décrite comme régulateur positif de la voie Wnt, notamment en interagissant et en phosphorylant directement Dv1. (Sun et al, 2001). Un autre mécanisme, régulant négativement la voie Wnt via Dv1 a été décrite. En effet la protéine I Dax (Inhibition of Dv1 Axin complex) interagit avec le domaine PDZ de Dv1 et inhibe la formation du complexe Dv1/Axine. (Hino et al, 2001). De même une nouvelle protéine nommée Axam (A Xin Molecule) entre en compétition avec Dv1 pour se lier à l'axine, sans empêcher GSK-3b, â caténine ou PP2A de s'y lier. Ces deux protéines empêchent ainsi Dv1 d'inhiber la phosphorylation de la â caténine par GSK-3b (Kadoya et al, 2000). Il se pourrait que le signal Wnt lève l'inhibition pour Axam et I Dax de la liaison de Dv1 à Axine (Sun et a/, 2001). 2.3.4. Régulation de la 3 â - caténine par Frat-1 En absence de stimulation de la voie Wnt, l'axine se lie à la GSK-3b, â caténine et APC, permettant ainsi à GSK-3b de phosphoryler la 3 â caténine. Dv1 se fixe au complexe en interagissant avec l'axine, mais ne parait pas jouer un rôle important en absence de signal Wnt. Dv1 se fixe également à un peptide nommé Frat-1 (Frequently Rerrangedin AdvandT Cell Lymphonas I). La voie Wnt mène à la déstabilisation de la GSK-3b de l'axine avec l'aide de Dv1 et Frat-1. Ceci suggère que dv1 serait capable de recruter Frat-1 au niveau du complexe comprenant l'axine. Ainsi, il pourrait agir avec la GSK-3b après stimulation par Wnt. (Li et a!, 1999). La protéine GBP homologue de Frat-1 chez le xénope a été décrite pour etre capable d'inhiber la phosphorylation et la dégradation de la â caténine in vitro (Salic et a!, 2000). Bien que Frat-1 contribue au développement du cancer dans un modèle de souris transgénique, son rôle dans les cancers humains n'est pas bien documenté. 2.3.5. Autres voies de régulation La compétition entre les différents partenaires de la â caténine participe à sa régulation. Par exemple, la surexpression des cadhérines provoque le recrutement de la majorité de la â caténine au niveau des jonctions adhérentes, ceci réduit sa capacité à se complexer avec les facteurs de transcription Tcf/LEF, et inhibe alors la transcription méfiée par la â caténine. (Orsulic et a!, 1999). L'effet inverse est obtenu en sur exprimant le facteur Tcf (Simcha et a!, 1998). L'état de cancérisation pourrait donc dépendre du rapport cadhérines/Tcf/LEF. Les interactions â caténine /cadhérines entrent également en compétition avec le complexe de dégradation (APC, Axine....) protégeant ainsi la â caténine, impliquée dans l'adhésion cellulaire, de la dégradation par le protéasome. 2.4. Mécanismes de dégradations de la â carénine 2.4.1. Le protéasome Le complexe formé autour de l'axine mène à la dégradation de la â carénine par le protéasome à l'aide de la kinase GSK-3â. En effet, la â caténine posséde une séquence consensus DSGXXS dont les phosphosérines (S33 et S37) sont reconnues par la â-TrCP. (Kitagawa et a!, 1999). L'ubiquitination des protéines requière l'action conjuguée de trois enzymes qui forment une cascade de réactions de transfert d'ubiquitine ligase : une enzyme d'activation E1, une enzyme de conjugaison E2 et une ubiquitine ligase E3 (Kitagawa et a!, 1999). Directement après avoir été phosphorylée par GSK-3â, la â caténine est prise en charge par â TrCP qui catalyse son ubiquitination. Le protéasome 26S va alors pouvoir la reconnaître et la dégrader. Â-TrCP est lui-même un des gènes cibles de la voie Wnt. Ainsi, plus les gènes cibles de Wnt sont activés, plus il y aura de â-TrCP pour dégrader la â caténine. C'est une boucle de rétrocontrôle négatif. Des résultats récents montrent que siah-1 (homologue humain du Drosophilia Sevenln Absentia), protéine dont l'expression est sous le contrôle de P53 et qui est impliqué dans l'arrêt du cycle cellulaire, a également la capacité de dégrader la â caténine. Cette dégradation est indépendante de la phosphorylation de la â caténine par GSK-3 â. Elle ne nécessite pas la protéine â-TrCP servant de récepteur à la â caténine phosphorylée et constituant le premier signal pour l'ubiquination et la dégradation de la â caténine (Liu et a!, 2001). Cependant, une autre protéine, Eb1 est utilisée par siah-1 dans le but d'ubiquitiner et de dégrader la â caténine (Fig18) (Matsuzawa et Reed, 2001).
Figure 18 : dégradation de la béta
caténine par le protéasome Conclusion ConclusionUne surveillance biologique soutenue incluant le dosage de l'ACE permet de prédire la survenue de récidive avec une avance de quelques semaines à quelques mois sur le diagnostic clinique ou radiologique et permet aussi le diagnostic des récidives à un stade où l'opérabilité est meilleure. Cependant, il n'existe pas à l'heure actuelle, à l'échelle d'une population de malades suivis, de bénéfice démontré, en termes de survie, résultant de cette avance au diagnostic et de cette meilleure opérabilité. L'inclusion du dosage de l'ACE dans la surveillance des cancers du côlon varie selon les comités d'experts. Les options sont les suivantes : pas de surveillance de l'ACE en dehors d'essais thérapeutiques ou surveillance régulière de l'ACE chez les patients en état de supporter une thérapeutique autre que symptomatique. En l'absence de consensus, la surveillance régulière des concentrations sériques de l'ACE après chirurgie à visée curative d'un cancer du côlon n'est pas recommandable de façon systématique. En tout état de cause, elle n'est envisageable que chez les sujets en état de supporter une thérapeutique autre que symptomatique en cas de documentation d'une récidive tumorale. Dans tous les cas, la place du dosage des marqueurs tumoraux dans le suivi des patients traités devra être réévaluée à la publication de nouvelles études randomisées et quand de nouvelles techniques d'imagerie (tomographie à émission de positons utilisant le 18 Fluorodéoxy-glucose, scanner hélicoïdal, etc.) et de chirurgie (traitement percutané des métastases, etc.) seront validées ou plus largement utilisées et que les thérapeutiques systémiques des cancers auront gagné en efficacité. Il semble clair que la beta-caténine puisse être définie comme un oncogène jouant un rôle important dans la progression tumorale colique. Elle est constitutivement activée très précocement au cours de la carcinogenèse par des altérations géniques qui peuvent soit l'affecter directement, soit toucher des protéines impliquées dans la régulation de sa dégradation, telles que APC ou GSK3beta Son activation constitutive conduit à l'expression dérégulée de gènes cibles impliqués dans des processus cellulaires tels que la prolifération ou l'invasion qui jouent un rôle important dans la progression tumorale. Références bibliographiques (1), www.chu-rouen.fr/ssf/phenobioch/digestion.html Agence nationale d'accréditation et d'évaluation en santé (Anaes), éd. Conférence de consensus Prévention, dépistage et prise en charge des cancers du côlon [On-Line]. Paris : Anaes ; 29-30 janvier 1998. Available : URL : http://www.anaes.fr. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMB O 1 1997 ; 16 : 3797-804. Adam R, Bismuth H, Castaing D, Waechter F, Navarro F, Abascal A, et al. Repeat hepatectomy for colorectal liver metastases. Ann Surg 1997 ; 225 : 51-60. Adenis A, Conroy T, Lasser P, Merrouche Y, Monges G, Rivoire M, et al. Standards, Options et Recommandations pour la prise en charge des patients atteints de cancer du colon. In : Fédération nationale des centres de lutte contre le cancer, éd. Recommandations pour la pratique clinique en cancérologie [cédérom]. 2e ed. Paris : FNCLCC, John Libbey Eurotext ; 1998. Standards, Options et Recommandations. Agence du Médicament. Controle de qualite national. Agence du médicament, éd. Saint Denis : 1997. Agence nationale d'accréditation et évaluation en santé (Anaes). Marqueurs seriques dans les cancers du sein et les cancers colorectaux. Paris : Anaes ; 1997 (Recommandations et References medicales). Allen-Mersh TG, Kemeny N, Niedzwiecki D, Shurgot B, Daly JM. Significance of a fall in serum CEA concentration in patients treated with cytotoxic chemotherapy for disseminated colorectal cancer. Gut 1987 ; 28 : 1625-9. Alvarez JA, Marin J, Jover JM, Fernandez R, Fradejas J, Moreno M. Sensitivity of monoclonal antibodies to carcinoembryonic antigen, tissue polypeptide antigen, alpha-fetoprotein, carbohydrate antigen 50, and carbohydrate antigen 19-9 in the diagnosis of colorectal adenocarcinoma. Dis Colon Rectum 1995 ; 38 : 35-42. Arnaud JP, Thibaud D, Leguillou A, Bergamaschi R, Adloff M. A prospective study of current diagnostic procedures for assessment of hepatic metastases in colorectal cancers. Eur J Surg Oncol 1987 ; 13 : 355-8. Assersohn L, Norman A, Cunningham D, Benepal T, Ross PJ, Oates J. Influence of metastatic site as an additional predictor for response and outcome in advanced colorectal carcinoma. Br J Cancer 1999; 79: 1800-5. Avital S, Haddad R, Troitsa A, Kashtan H, Brazovsky E, Gitstein G, et al. Radioimmunoguided surgery for recurrent colorectal cancer manifested by isolated CEA elevation. Cancer 2000 ; 89 : 1692-8. Baeg GH, Matsumine A, Kuroda T, et al. The tumor suppressor gene product APC blocks cell cycle progression from G0/G1 to S phase. EMB O J 1995 ; 14 : 5618-25. Bakalakos EA, Burak WE, Young DC, Martin EW. Is carcino-embryonic antigen useful in the follow-up management of patients with colorectal liver metastases ? Am J Surg 1999 ; 177 : 2-6. Ballesta AM, Molina R, Filella X, Jo J, Gimenez N. Carcinoembryonic antigen in staging and follow-up of patients with solid tumors. Tumour Biol 1995 ; 16 : 32-41. Baron O, Amini M, Duveau D, Despins P, Sagan CA, Michaud JL. Surgical resection of pulmonary metastases from colorectal carcinoma : five-year survival and main prognostic factors. Eur J Cardio Thoracic Surg 1996 ; 10 : 347-51. Bast RC, Ravdin P, Hayes DF, Bates S, Fritsche H, Jessup JM, et al. 2000 update of recommendations for the use of tumor markers in breast and colorectal cancer : clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol 2001 ; 19 : 1865-78. Basuyau JP, Blanc-Vincent MP, Bidart JM, Daver A, Deneux L, Eche N, et al. Standards, Options et Recommandations : marqueurs tumoraux sériques et cancer du sein. Bull Cancer 2000 ; 87 : 723-37. Beauchemin N, Draber P, Dveksler G, Gold P, Gray-Owen S, Grunert F, et al. Redefined nomenclature for members of the carcinoembryonic antigen family. Exp Cell Res 1999 ; 252 : 243-9. Beets G, Penninckx F, Schiepers C, Filez L, Mortelmans L, Kerremans R, et al. Clinical value of whole-body positron emission tomography with [18F]fluorodeoxyglucose in recurrent colorectal cancer. Br J Surg 1994 ; 81 : 1666-70. Behbehani AE, Dashti H, Hussain T, Szymendera JJ. Five-year survival versus initial concentrations and longitudinal serum CEA patterns in patients with colorectal carcinoma. Nutrition 1995 ; 11 (suppl.) : 8. Behrens J, Jerchow BA, Würtele M, Grimm J, Asbrand C, Wirtz R, et al. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science 1998 ; 280 : 596-9. Behrens J, van Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996 ; 382 : 638-42. Benchimol S, Fuks A, Jothy S, Beauchemin N, Shirota K, Stanners CP. Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell 1989 ; 57 : 327-34. Bergamaschi R, Arnaud JP. Routine compared with nonscheduled follow-up of patients with « curative » surgery for colorectal cancer. Ann Surg Oncol 1996 ; 3 : 464- 9. Bhatavdekar JM, Patel DD, Giri DD, Karelia NH, Vora HH, Ghosh N, et al. Comparison of plasma prolactin and CEA in monitoring patients with adenocarcinoma of colon and rectum. Br J Cancer 1992 ; 66 : 977-80. Bodmer W, Bailey C, Bodmer J, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 1987 ; 328 : 614-6. Boey J, Cheung HC, Lai CK, Wong J. A prospective evaluation of serum carcinoembryonic antigen (CEA) levels in the management of colorectal carcinoma. World I Surg 1984 ; 8 : 279-86. Bonfrer JM. Working Group on Tumor Marker Criteria (WGTMC). [Letter] Tumour Biol 1990 ; 11 : 287-8. Bonithon-Kopp C; Kronborg O; Giacosa A; Räth U; Faivre J Calcium and fibre supplementation in prevention of colorectal adenoma recurrence: a ..., 2000. Bonneton C, Larue L, Thiery JP. The APC gene product and colorectal carcinogenesis. C R Acad Sci Paris Sciences de la Vie 1996 ; 31 : 861-9. Bruinvels DJ, de Brauw LM, Kievit J, Habbema JD, van de Velde CJ. Attitudes towards detection and management of hepatic metastases of colorectal origin : a second look. HPB Surgery 1994 ; 8 : 115-22. Bruinvels DJ, Stiggelbout AM, Kievit J, van Houwelingen HC, Habbema JD, van de Velde CJ. Follow-up of patients with colorectal cancer : a meta-analysis. Ann Surg 1994 ; 219 : 174-82. Calmettes C, Moukhtar MS, Milhaud G. Tumours markers. Calcitonin and carcinoembryonic antigen in medullary carcinoma of the thyroid. Presse Med 1979 ; 8 : 3947-50. Canney PA, Wilkinson PM, James RD, Moore M. CA19-9 as a marker for ovarian cancer : alone and in comparison with CA125. Br I Cancer 1985 ; 52 : 131-3. Carl J, Bentzen SM, Norgaard-Pedersen B, Kronborg O. Modelling of serial carcinoembryonic antigen changes in colorectal cancer. Scand I Clin Lab Invest 1993 ; 53 : 751-5. Carlsson U, Stewenius J, Ekelund G, Leandoer L, Nosslin B. Is CEA analysis of value in screening for recurrences after surgery for colorectal carcinoma ? Dis Colon Rectum 1983 ; 26 : 369-73. Carpelan-Holmstrom M, Haglund C, Lundin J, Alfthan H, Stenman UH, Roberts PJ. Independent prognostic value of preoperative serum markers CA 242, specific tissue polypeptide antigen and human chorionic gonadotrophin beta, but not of carcinoembryonic antigen or tissue polypeptide antigen in colorectal cancer. Br I Cancer 1996 ; 74 : 925-9. Carpelan-Holmstrom M, Haglund C, Lundin J, Jarvinen H, Roberts P. Preoperative serum levels of CA 242 and CEA predict outcome in colorectal cancer. Eur I Cancer 1996 ; 32A : 1156-61. Carpelan-Holmstrom MA, Haglund CH, Roberts PJ. Differences in serum tumor markers between colon and rectal cancer : comparison of CA 242 and carcinoembryonic antigen. Dis Colon Rectum 1996 ; 39 : 799-805. Cases A, Filella X, Molina R, Ballesta AM, Lopez-Pedret J, Revert L. Tumor markers in chronic renal failure and hemodialysis patients. Nephron 1991 ; 57 : 183-6. Caspari R, Olshwang S, Friedl W, et al. Familial adenomatous polyposis : desmoid tumours and lack of ophtalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet 1995 ; 4 : 337-40. Chakrabarty S, Fan D, Varani J. Modulation of differentiation and proliferation in human colon carcinoma cells by transforming growth factor beta 1 and beta 2. Int .1 Cancer 1990 ; 46 : 493-9. Chakrabarty S, Tobon A, Varani J, Brattain MG. Induction of carcinoembryonic antigen secretion and modulation of protein secretion/expression and fibronectin/laminin expression in human colon carcinoma cells by transforming growth factor-beta. Cancer Res 1988 ; 48 : 4059-64. Chapman MA, Buckley D, Henson DB, Armitage NC. Preoperative carcinoembryonic antigen is related to tumour stage and long-term survival in colorectal cancer. Br J Cancer 1998 ; 78 : 1346-9. Chi KF, Jessup JM, Frazier ML. Predominant expression of mRNA coding for nonspecific cross-reacting antigen in colorectal carcinomas. Tumour Biol 1991 ; 12 : 298-308. Chu DZ, Ellenhorn JD, Paz IB, Wagman LD, Williams WL. CEA for monitoring colon cancer. JAMA 1994 ; 271 : 345-6. Cohen AM, Minsky BD, Schilsky RL. Colon cancer. In : De Vita VTJ, Hellman S, Rosenberg SA, eds. Cancer principle and practice of oncology. Philadelphia : Lippincott, 1993 ; 929-77. Cohen R, Zucchelli GC, Fraysse M, Pilo A, Rigault MY, Grillet S, et al. « Oncocheck » : an international external quality assessment scheme for immunoassays of tumor markers. Nucl Med Biol 1994 ; 21 : 483-93. Conroy T, Adenis A. Standards, Options et Recommandations pour la surveillance après traitement d'un cancer du côlon. Le groupe de travail SOR Cancer du Colon de la Fédération nationale des centres de lutte contre le cancer. Bull Cancer 1998 ; 85 : 152-9. Costanza ME, Das S, Nathanson L, Rule A, Schwartz RS. Proceedings : carcinoembryonic antigen. Report of a screening study. Cancer 1974 ; 33 : 583-90. Crawford HC, Fingleton BM, Rudolph-Owen LA, Heppner Goss KJ, Rubinfeld B, Polakis P, et al. The metalloproteinase matrilysin is a target of betacatenin transactivation in intestinal tumors. Oncogene 1999 ; 18 : 2883-91. Crawford HC, Fingleton BM, Rudolph-Owen LA, Heppner Goss KJ, Rubinfeld B, Polakis P, et al. The metalloproteinase matrilysin is a target of betacatenin transactivation in intestinal tumors. Oncogene 1999 ; 18 : 2883-91. Denstman F, Rosen L, Khubchandani IT, Sheets JA, Stasik JJ, Riether RD. Comparing predictive decision rules in postoperative CEA monitoring. Cancer 1986 ; 58 : 2089-95. Desch CE, Benson AB, Smith TJ, Flynn PJ, Krause C, Loprinzi CL, et al. Recommended colorectal cancer surveillance guidelines by the American Society of Clinical Oncology. J Clin Oncol 1999 ; 17 : 1312-21. Deugnier YM, Rabot AF, Guyader D, Moirand R, Turlin B, Boucher E, et al. Serum increase and liver overexpression of carbohydrate 19.9 antigen in patients with genetic haemochromatosis. Gut 1994 ; 35 : 1107-11. Doerr RJ, Herrera L, Abdel-Nabi H. In-111 CYT-103 monoclonal antibody imaging in patients with suspected recurrent colorectal cancer. Cancer 1993 ; 71 (suppl. 12) : 4241-7. Dorudi D, Sheffield JP, Poulsom R, Northover J, Hart IR. E-cadherin expression in colorectal cancer. An immunocytochemical and in situ hybridization study. Am J Pathology 1993 ; 142 : 981-6. Durand DV. Marqueurs tumoraux et dépistage des cancers : les raisons d'un échec. Rev Prescrire 1990 ; 10 : 156-61. Erbil KM, Jones JD, Klee GG. Use and limitations of serum total and lipid-bound sialic acid concentrations as markers for colorectal cancer. Cancer 1985 ; 55 : 404-9. Esteban JM, Colcher D, Sugarbaker P, Carrasquillo JA, Bryant G, Thor A, et al. Quantitative and qualitative aspects of radiolocalization in colon cancer patients of intravenously administered MAb B72.3. Int J Cancer 1987 ; 39 : 50-9. Fagotto F, Gluck U, Gumbiner BM. Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of beta-catenin. Curr Biol 1998 ; 8 : 181-90. Fervers B, Bonichon F, Demard F, Heron JF, Mathoulin S, Philip T, RenaudSalis JL, Toma C, Bey P. Méthodologie de développement des Standards, Options et Recommandations diagnostiques et thérapeutiques en cancérologie. Bull Cancer 1995 ; 82 : 761-7. Filella X, Molina R, Grau JJ, Pique JM, Garcia-Valdecasas JC, Astudillo E, et al. Prognostic value of CA 19.9 levels in colorectal cancer. Ann Surg 1992 ; 216 : 55-9. Filella X, Molina R, Pique JM, Garcia-Valdecasas JC, Grau JJ, Novell F, et al. Use of CA 19-9 in the early detection of recurrences in colorectal cancer : comparison with CEA. Tumour Biol 1994 ; 15 : 1-6. Filella X, Molina R, Pique JM, Grau JJ, Garcia-Valdecasas JC, Biete A, et al. CEA as a prognostic factor in colorectal cancer. Anticancer Res 1994 ; 14 : 705-8. Flanagan FL, Dehdashti F, Ogunbiyi OA, Kodner IJ, Siegel BA. Utility of FDG-PET for investigating unexplained plasma CEA elevation in patients with colorectal cancer. Ann Surg 1998 ; 227 : 319-23. Fletcher RH. Carcinoembryonic antigen. Ann Intern Med 1986 ; 104 : 66-73. Fodde R, Edelmann W, Yang K, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci USA 1994 ; 91 : 8969-73. Foulkes WD. A tale of four syndromes : familial adenomatous polyposis, attenuated APC and Turcot syndrome. Q J Med 1995 ; 88 : 853-63. Fountzilas G, Gossios K, Zisiadis A, Svarna E, Skarlos D, Pavlidis N. Prognostic variable in patients with advanced colorectal cancer treated with fluorouracil and leucovorin-based chemotherapy. Med Ped Oncol 1996 ; 26 : 305-17. Friedl W, Meuschel S, Caspari R, Lamberti C, Krieger S, Sengteller M, et al. Attenuated familial adenomatous polyposis due to a mutation in the 3' part of the APC gene : a clue for understanding the function of the APC protein. Hum Genet 1996 ; 97 : 579-84. Fukuta S, Magnani JL, Gaur PK, Ginsburg V. Monoclonal antibody CC3C195, which detects cancer-associated antigens in serum, binds to the human Lea blood group antigen and to its sialylated derivative. Arch Biochem Biophys 1987 ; 255 : 214-6. Gadducci A, Ferdeghini M, Prontera C, Moretti L, Mariani G, Bianchi R, et al. The concomitant determination of different tumor markers in patients with epithelial ovarian cancer and benign ovarian masses : relevance for differential diagnosis. Gynecol Oncol 1992 ; 44 : 147-54. Gangopadhyay A, Lazure DA, Kelly TM, Thomas P. Purification and analysis of an 80-kDa carcinoembryonic antigen-binding protein from Kupffer cells. Arch Biochem Biophys 1996 ; 328 : 151-7. Gangopadhyay A, Bajenova O, Kelly TM, Thomas P. Carcinoembryonic antigen induces cytokine expression in Kuppfer cells : implications for hepatic metastasis from colorectal cancer. Cancer Res 1996 ; 56 : 4805-10. Gangopadhyay A, Lazure DA, Thomas P. Adhesion of colorectal carcinoma cells to the endothelium is mediated by cytokines from CEA stimulated Kupffer cells. Clin Exp Metastasis 1998 ; 16 : 703-12. Girard P, Ducreux M, Baldeyrou P, Rougier P, Le Chevalier T, Bougaran J, et al. Surgery for lung metastases from colorectal cancer : analysis of prognostic factors. J Clin Oncol 1996 ; 14 : 2047-53. Gold P, Freedman SO. Demonstration of tumor-specific antigens in human colon carcinoma by immunological tolerance and absorption techniques. J Exp Me 1965 ; 122 : 467-81. Goslin R, Steele G, Jr., Macintyre J, Mayer R, Sugarbaker P, Cleghorn K, et al. The use of preoperative plasma CEA levels for the Stratification of patients after curative resection of colorectal cancers. Ann Surg 1980 ; 192 : 747-51. Goslin R, O'Brien MJ, Steele G, Mayer R, Wilson R, Corson JM, et al. Correlation of Plasma CEA and CEA tissue staining in poorly differentiated colorectal cancer. Am J Med 1981 ; 71 : 246-53. Graffner H, Hultberg B, Johansson B, Moller T, Petersson BG. Detection of recurrent cancer of the colon and rectum. J Surg Oncol 1985 ; 28 : 156-9. Grassi J. Interférences dues aux anticorps anti-immunoglobulines, un poison pour tous les dosages immunologiques. I. Origine des anticorps antiimmunoglobulines et mécanismes des interférences. Immunoanal Biol Spec 1994 ; 9 : 60-7. Grassi J. Interférences dues aux anticoprs anti-immunoglobulines, un poison pour tous les dosages immunologiques. II. Interférences, mises en évidence et suppression. Immunoanal Biol Spec 1994 ; 9 : 124-33. Greiner JW, Guadagni F, Goldstein D, Borden EC, Ritts RE, Witt P, et al. Evidence for the elevation of serum carcinoembryonic antigen and tumor- associated glycoprotein-72 levels in patients administered interferons. Cancer Res 1991 ; 51 : 4155-63. Grimm T, Johnson JP. Ectopic expression of carcinoembryonic antigen by a melanoma cell leads to changes in the transcription of two additional cell adhesion molecules. Cancer Res 1995 ; 55 : 3254-7. Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis gene. Cell 1991 ; 66 : 589-600. Guadagni F, Witt PL, Robbins PF, Schlom J, Greiner JW. Regulation of carcinoembryonic antigen expression in different human colorectal tumor cells by interferon-gamma. Cancer Res 1990 ; 50 : 6248-55. Gumbiner BM. Signal transduction of beta-catenin. Curr Opin Cell Biol 1995 ; 7 : 634-40. Haglund C, Lundin J, Kuusela P, Roberts PJ. CA 242, a new tumour marker for pancreatic cancer : a comparison with CA 19-9, CA 50 and CEA. Br J Cancer 1994 ; 70 : 487-92. Haglund C, Ylatupa S, Mertaniemi P, Partanen P. Cellular fibronectin concentration in the plasma of patients with malignant and benign diseases : a comparison with CA 19-9 and CEA. Br J Cancer 1997 ; 76 : 777-83. Halmilton SR, Liu B, Parsons RE, et al. The molecular basis of Turcot's syndrome (see comments). N Engl J Med 1995 ; 332 : 839-47. Hamm CM, Cripps C. Carcinoembryonic antigen in metastatic colorectal cancer. Clin Investig Med 1998 ; 21 : 186-91. Hammarström S. Chemistry and immunology of CEA, CA 19-9 and CA-50. In & Holmgren J, ed. Tumor marker antigens. Lund : Studentlitteratur ed. 1985 : 34-51. Hammarström S, Shively JE, Paxton RJ, Beatty BG, Larsson A, Ghosh R, et al. Antigenic sites in carcinoembryonic antigen. Cancer Res 1989 ; 49 : 4852-8. Hammarström S. The carcinoembryonic antigen (CEA) family : structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 1999 ; 9 : 67-81. Hanazaki K, Kawamura N, Sodeyama H, Wakabayashi M, Yokoyama S, Sode Y, et al. Carcinoembryonic antigen and intra-arterial chemotherapy response of liver metastases. Hepato-Gastroenterology 1998 ; 45 : 462-7. Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMB 0 J 1999 ; 18 : 5931-42. Harrison LE, Guillem JG, Paty P, Cohen AM. Preoperative carcinoembryonic antigen predicts outcomes in node-negative colon cancer patients : a multivariate analysis of 572 patients. J Am Coll Surg 1997 ; 185 : 55-9. Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P. Downregulation of beta-catenin by human axin and its association with the APC tumor suppressor, beta-catenin and GSK3beta. Curr Biol 1998 ; 8 : 573-81. Hashino J, Fukuda Y, Oikawa S, Nakazato H, Nakanishi T. Metastatic potential of human colorectal carcinoma SW1222 cells transfected with cDNA encoding carcinoembryonic antigen. Clin Exp Metastasis 1994 ; 12 : 324-8. Hayashi S, Rubinfeld B, Souza B, Polakis P, Wieschaus E, Levine AJ. A drosophila homolog of the tumor suppressor gene adenomatous polyposis coli down-regulates beta catenin but its zygotic expression is not essential for the regulation of armadillo. Proc Natl Acad Sci USA 1997 ; 94 : 242-7. He TC, Spaks AB, Rago C, Hermeking H, Zawel L, Da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science 1998 ; 281 : 1509-12. Heasman J., et al. 1994. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell 79 : 791-803. Herlyn M, Steplewski Z, Herlyn D, Koprowski H. Colorectal carcinoma-specific antigen : detection by means of monoclonal antibodies. Proc Natl Acad Sci USA 1979 ; 76 : 1438-52. Hermitson ML, Gordon JI. In vivo analysis of cadherin function in the mouse intestinal epithelium : essential roles in adhesion, maintenance of differentiation and regulation of programmed cell death. J Cell Biol 1995 ; 129 : 489-506. Ho JJ, Kim YS. Serological pancreatic tumor markers and the MUC1 apomucin. Pancreas 1994 ; 9 : 674-91. Hohenberger P, Schlag PM, Gerneth T, Herfarth C. Pre- and postoperative carcinoembryonic antigen determinations in hepatic resection for colorectal metastases : predictive value and implications for adjuvant treatment based on multivariate analysis. Ann Surg 1994 ; 219 : 135-43. Hostetter RB, Augustus LB, Mankarious R, Chi KF, Fan D, Toth C, et al. Carcinoembryonic antigen as a selective enhancer of colorectal cancer metastasis. J Natl Cancer Inst 1990 ; 82 : 380-5. Huber O, Korn R, McLaughlin J, Ohsugi M, Herrman BG, Kemler R. Nuclear Localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev 1996 ; 59 : 3-10. Hughes K, Pinsky CM, Petrelli NJ, Moffat FL, Patt YZ, Hammershaimb L, et al. Use of carcinoembryonic antigen radioimmunodetection and computed tomography for predicting the resectability of recurrent colorectal cancer. Ann Surg 1997 ; 226 : 621-31. Hülsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with beta -catenin and cytoskeleton. I Cell Biol 1994 ; 127 : 2061-9. Iemura K, Moriya Y. A comparative analysis of the serum levels of NCC-ST439, CEA and CA19-9 in patients with colorectal carcinoma. Eur I Surg 0ncol 1993 ; 19 : 439-42. Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSKbeta and beta-catenin and promotes GSK3beta-dependent phosphorylation of beta-catenin. EMB 0 1 1998 ; 17 : 1371-84. Iwao K, Nakamori S, Kameyama M, Imaoka S, Kinoshita M, Fukui T, et al. Activation of the beta-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res 1998 ; 58 : 1021-6. Jen J, Powell SM, Papadopoulos N, Smith KJ, Hamilton SR, Vogelstein B, et al. Molecular determinants of dysplasia in colorectal lesions. Cancer Res 1994 ; 54 : 5523-6. Johnson JP. Cell adhesion molecules of the immunoglobulin supergene family and their role in malignant transformation and progression to metastatic disease. Cancer Metastasis Rev 1991 ; 10 : 11-22. Jothy S, Yuan SY, Shirota K. Transcription of carcinoembryonic antigen in normal colon and colon carcinoma. In situ hybridization study and implication for a new in vivo functional model. Am .1 Pathol 1993 ; 143 : 250-7. Kammerer R, von Kleist S. CEA expression of colorectal adenocarcinomas is correlated with their resistance against LAK-cell lysis. Int .1 Cancer 1994 ; 57 : 341-7. Kammerer R, von Kleist S. The carcinoembryonic antigen (CEA) modulates effector-target cell interaction by binding to activated lymphocytes. Int .1 Cancer 1996 ; 68 : 457-63. Kashiwaba M, Tamura G, Ishida M. Aberrations of the APC gene in primary breast carcinomas. .1 Cancer Res Clin Oncol 1994 ; 120 : 727-31. Kawaharata H, Hinoda Y, Itoh F, Endo T, Oikawa S, Nakazato H, et al. Decreased sensitivity of carcinoembryonic antigen cDNA-transfected cells to adriamycin. Int .1 Cancer 1997 ; 72 : 377-82. Kemeny MM, Sugarbaker PH, Smith TJ, Edwards BK, Shawker T, Vermess M, et al. A prospective analysis of laboratory tests and imaging studies to detect hepatic lesions. Ann Surg 1982 ; 195 : 163-7. Kemler R. 1993. From cadherins to catenins : cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet 9 : 317-321. Kim JC, Koo KH, Kim BS, Park KC, Bicknell DC, Bodmer WF. Carcinoembryonic antigen may function as a chemo-attractant in colorectal-carcinoma cell lines. Int .1 Cancer 1999 ; 82 : 880-5. Kinzler KW, Nilbet MC, Su LK, et al. Identification of FAP locus gene from chromosome 5q21. Science 1991 ; 253 : 661-4. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996 ; 87 : 159-70. Koch M, Washer G, Gaedke H, McPherson TA. Carcinoembryonic antigen : usefulness as a postsurgical method in the detection of recurrence in Dukes stages B2 and C colorectal cancers. J Natl Cancer Inst 1982 ; 69 : 813-5. Koh TJ, Docray GJ, Varro A, Cahill RJ, Dangler CA, Fox JG, et al. Overexpression of glycine-extended gastrin in transgenic mice results in increased colonic proliferation. J Clin Invest 1999 ; 103 : 1119-26. Koh TJ, Bulitta CJ, Fleming JV, Docray GJ, Varro A, Wang TC. Gastrin is a target of the beta-catenin/TCF-4 growth-signaling pathway in a model of intestinal polyposis. J Clin Invest 2000 ; 106 : 533-9. Kohn J. The value of apparent half life assay of a1-fetaprotein in the management of testicular teratoma. In : Lehmann FG, ed. Carcino-embryonic proteins : chemistry, biology, clinical applications. vol. II. Amsterdam : Elsevier/North-Holland Biomedical Press. 1979 ; 932 p. Koprowski H, Steplewski Z, Mitchell K, Herlyn M, Herlyn D, Fuhrer P. Colorectal carcinoma antigens detected by hybridoma antibodies. Somatic Cell Genet 1979 ; 5 : 957-71. Korenaga D, Saeki H, Mawatari K, Orita H, Maekawa S, Ikeda T, et al. Serum carcinoembryonic antigen concentration doubling time correlates with tumor biology and life expectancy in patients with recurrent gastrointestinal carcinoma [published erratum appears in Arch Surg 1997 Jun ; 132 : 682]. Arch Surg 1997 ; 132 : 188-94. Korinek V., et al. 1997. Constitutive transcriptional activation by a beta-cateninTcf complex in APC/ colon carcinoma. Science 275 : 1784-1787. Kornek G, Pfeffel F, Schenk T, Bauer J, Groz S, and Scheithauer W. TPS for monitoring of response to chemotherapy in gastrointestinal (GI) cancer. Proc Annu Meet Am Assoc Cancer Res 1992 ; 33 : A2107. Kuusela P, Haglund C, Roberts PJ. Comparison of a new tumour marker CA 242 with CA 19-9, CA 50 and carcinoembryonic antigen (CEA) in digestive tract diseases. Br I Cancer 1991 ; 63 : 636-40. Kuusela P, Jalanko H, Roberts P, Sipponen P, Mecklin JP, Pitkanen R, et al. Comparison of CA 19-9 and carcinoembryonic antigen (CEA) levels in the serum of patients with colorectal diseases. Br I Cancer 1984 ; 49 : 135-9. Larsen JC. Les perversions du couple beta -caténine/protéine APC dans la cancérogenèse colique. Bull Cancer 1997 ; 84 : 772-3. Lassau N, Leclere J, Elias D, Lasser P. Role of imaging in abdominopelvic follow-up after resection of colorectal cancer. I Chir (Paris) 1997 ; 134 : 51-8. Leconte A, Garambois V, Ychou M, Robert B, Pourquier D, Terskikh A, et al. Involvement of circulating CEA in liver metastases from colorectal cancers reexamined in a new experimental model. Br I Cancer 1999 ; 80 : 1373-9. Lejeune S, Huguet EL, Hamby A, Poulsom R, Harris AL. Wnt5a cloning, expression, and up-regulation in human primary breast cancers. Clin Cancer Res 1995 ; 1 : 215-22. Leppert M, Burt R, Hughes J, et al. Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N Engl I Med 1990 ; 322 : 904-8. Levy DB, Smith KJ, Beaze-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res 1994 ; 54 : 5953-8. Létourneau S, Beauchemin N. Rôles des antigènes carcino-embryonnaires dans la cancérisation et la progression tumorale. medecine/sciences 1997 ; 13 : 483-91. Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin-Tcf signaling. Nature Genet 2000 ; 26 : 146-7. Lindmark G, Bergstrom R, Pahlman L, Glimelius B. The association of preoperative serum tumour markers with Dukes' stage and survival in colorectal cancer. Br I Cancer 1995 ; 71 : 1090-4. Lucha PA, Jr., Rosen L, Olenwine JA, Reed JF, III, Riether RD, Stasik JJ, et al. Value of carcinoembryonic antigen monitoring in curative surgery for recurrent colorectal carcinoma. Dis Colon Rectum 1997 ; 40 : 145-9. Lunde OC, Havig O. Clinical significance of carcinoembryonic antigen (CEA) in patients with adenocarcinoma in colon and rectum. Acta Chir Scand 1982 ; 148 : 189- 93. Mach JP, Vienny H, Jaeger P, Haldemann B, Egely R, Pettavel J. Long-term follow-up of colorectal carcinoma patients by repeated CEA radioimmunoassay. Cancer 1978 ; 42 (suppl. 3) : 1439-47. Madara JL, Pappenheimer JR. Structural basis for physiological regulation of paracellular pathways in intestinal , 1991 Mar;100(3):719-724. [PubMed]. Magnani JL, Nilsson B, Brockhaus M, Zopf D, Steplewski Z, Koprowski H, et al. A monoclonal antibody-defined antigen associated with gastrointestinal cancer is a ganglioside containing sialylated lacto-N-fucopentaose II. I Biol Chem 1982 ; 257 : 14365-9. Maillart PH, Gamelin E, Goudier MJ, Gesta P, Burtin P, Danquechain-Dorval E, et al. Plasma kinetic of 5-fluorouracil : a high correlation with tumor-marker evolution and response to the treatment in metastatic colorectal cancer. Ann Oncol 1992 ; 3 (suppl. 5) : 115. Makela JT, Laitinen SO, Kairaluoma MI. Five-year follow-up after radical surgery for colorectal cancer. Results of a prospective randomized trial. Arch Sur 1995 ; 130 : 1062-7. Maldonado A, Sancho F, Cerdan J, Lozano A, Mohedano N, Jimenez J, et al. FDG-PET in the detection of recurrence in colorectal cancer based on rising CEA level : experience in 72 patients. Clin Positron Imaging 2000 ; 3 : 170. Martin EW, James KK, Hurtubise PE, Catalano P, Minton JP. The use of CEA as an early indicator for gastrointestinal tumor recurrence and second-look procedures. Cancer 1977 ; 39 : 440-6. Martin EW, Minton JP, Carey LC. CEA-directed second-look surgery in the asymptomatic patient after primary resection of colorectal carcinoma. Ann Surg 1985 ; 202 : 310-7. Matsumine A., et al. 1996. Binding of APC to the human homolog of the drosophila disc large tumor suppressor protein. Science 272 : 1020-1023. Matsumine A, Ogai A, Senda T, et al. Binding of APC to the human homolog of the Drosophila discs large tumor suppressor protein. Science 1996 ; 272 : 1020-3. Matsunaga A, Kuroki M, Higuchi H, Arakawa F, Takakura K, Okamoto N, et al. Antigenic heterogeneity of carcinoembryonic antigen in the circulation defined by monoclonal antibodies against the carbohydrate moiety of carcinoembryonic antigen and closely related antigens. Cancer Res 1987 ; 47 : 56-61. McCall JL, Black RB, Rich CA, Harvey JR, Baker RA, Watts JM, et al. The value of serum carcinoembryonic antigen in predicting recurrent disease following curative resection of colorectal cancer. Dis Colon Rectum 1994 ; 37 : 875-81. Mercier M, Morin JF. Valeur diagnostique d'un test : applications cliniques. In : Besnard JC, Morin JF, eds. Outils statistiques en immuno-analyse. Paris : Editions Nucléon ; 1997 ; 327-30. Minton JP, Hoehn JL, Gerber DM, Horsley JS, Connolly DP, Salwan F, et al. Results of a 400-patient carcinoembryonic antigen second-look colorectal cancer study. Cancer 1985 ; 55 : 1284-90. Miyaki M, Konishi M, Kikuchi-Yanoshita R, et al. Characteristics of the adenomatous polyposis gene in colorectal tumors. Cancer Res 1994 ; 54 : 3011-20. Miyoshi Y, Ando H, Nagase H, Nishisho I, Horii A, Miki Y, et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci USA 1992 ; 89 : 4452-6. Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, et al. Somatic mutations of the APC gene in colorectal tumors : mutation cluster region in the APC gene. Hum Mol Genet 1992 ; 1 : 229-33. Mlika-Cabanne N, Bellet D. Serum markers for breast and colorectal cancers. Working group reunited by the National Health Agency for Accreditation and Evaluation (NHAAE). Gastroenterol Clin Biol 1998 ; 22 : 442-57. Moertel CG, Fleming TR, Macdonald JS, Haller DG, Laurie JA, Tangen C. An evaluation of the carcinoembryonic antigen (CEA) test for monitoring patients with resected colon cancer. JAMA 1993 ; 270 : 943-7. Moertel CG, O'Fallon JR, Go VL, O'Connell MJ, Thynne GS. The preoperative carcinoembryonic antigen test in the diagnosis, staging, and prognosis of colorectal cancer. Cancer 1986 ; 58 : 603-10. Molitch ME, Schwartz S, Mukherji B. Is prolactin secreted ectopically ? Am I Med 1981 ; 70 : 803-7. Mora J, Queralto JM. Within-subject variation of carcinoembryonic antigen in colorectal cancer - application of reference of change values and individual reference ranges to patient follow-up. Eur I Clin Chem Clin Biochem 1997 ; 35 : 453-7. Morin JF. Notion de probabilité. In : Besnard JC, Morin JF, eds. Outils statistiques en immuni-analyse. Paris : Editions Nucléon. 1997 ; 233 p. Morin P.J., et al. 1997. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275 : 1787-1790. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997 ; 275 : 1787-90. Morin PJ, Vogelstein B, Kinzler KW. Apoptosis and APC colorectal tumorigenesis. Proc Natl Acad Sci USA 1996 ; 93 : 7950-4. Morin P, Sparks A, Korinek V, et al. Activation of beta -catenin-Tcf signaling in colon cancer by mutations in beta -catenin or APC. Science 1997 ; 275 : 1787-90. Munemitsu S., Albert I., Rubinfeld B., Polakis P. 1996. Deletion of an amino-terminal sequence stabilized beta -catenin in vivo and promotes hyperphosphorylation of the adenomatous polyposis coli tumor suppressor protein. Mol Cel Biol 16 : 4088-4094. Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta -catenin levels by adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Nat Acad Sci USA 1995 ; 92 : 3046-50. Nagase H, Nakamura Y. Mutations of the APC (adenomatous polyposis coli gene). Human Mutat 1993 ; 2 : 425-34. Nakayama T, Watanabe M, Teramoto T, Kitajima M. Prognostic values of serum CA 19-9 and CEA for colorectal cancer. Oncol Rep 1997 ; 4 : 822. Näthke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. I Cell Biol 1996 ; 134 : 165-79. Neumaier M, Paululat S, Chan A, Matthaes P, Wagener C. Biliary glycoprotein, a potential human cell adhesion molecule, is down-regulated in colorectal carcinomas. Proc Nat Acad Sci USA 1993 ; 90 : 10744-8. Niederhuber JE, Ensminger WD. Treatment of metastatic cancer to the liver. In : De Vita VTJ, Hellman S, Rosenberg SA, eds. Cancer principle and practice of oncology. Philadelphia : Lippincott ; 1993 : 2201-25. Nilsson O, Johansson C, Glimelius B, Persson B, Norgaard-Pedersen B, Andren-Sandberg A, et al. Sensitivity and specificity of CA242 in gastro-intestinal cancer. A comparison with CEA, CA50 and CA 19-9. Br J Cancer 1992 ; 65 : 215-21. Nishido J, Nakamura Y, Miyoshi Y, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991 ; 253 : 665-9. Noda M, Kusunoki M, Yanagi H, Yamamura T, Utsunomiya J. Serum carcinoembryonic antigen (CEA) correlates with the survival time during 5-FU hepatic arterial infusion chemotherapy for unresectable colorectal hepatic metastases. Int J Oncol 1996 ; 9 : 741-6. Nollau P, Prall F, Helmchen U, Wagener C, Neumaier M. Dysregulation of carcinoembryonic antigen group members CGM2, CD66a (biliary glycoprotein), and nonspecific cross-reacting antigen in colorectal carcinomas. Comparative analysis by northern blot and in situ hybridization. Am J Pathol 1997 ; 151 : 521-30. Nordlinger B, Guiguet M, Vaillant JC, Balladur P, Boudjema K, Bachellier P, et al. Surgical resection of colorectal carcinoma metastases to the liver. A prognostic scoring system to improve case selection, based on 1 568 patients. Association francaise de chirurgie. Cancer 1996 ; 77 : 1254-62. Northover JM. Carcinoembryonic antigen and recurrent colorectal cancer. Br J Surg 1985 ; 72 (suppl.) : S44-6. Northover J, Houghton J, Lennon T. CEA to detect recurrence of colon cancer. JAMA 1994 ; 272 : 31. Novis BH, Gluck E, Thomas P, Steele GD, Zurawski VR, Stewart R, et al. Serial levels of CA 19-9 and CEA in colonic cancer. J Clin Oncol 1986 ; 4 : 987-93. Nusse R., Varmus H.E. 1992. Wnt genes. Cell 69 : 1073-1087. Obrand DI, Gordon PH. Incidence and patterns of recurrence following curative resection for colorectal carcinoma. Dis Colon Rectum 1997 ; 40 : 15-24. Oda H, Nakatsusru Y, Ishikawa T. Somatic mutations of the APC gene in sporadic hepatoblastomas. Cancer Res 1996 ; 56 : 3320-3. Ohannesian DW, Lotan D, Thomas P, Jessup JM, Fukuda M, Gabius HJ, et al. Carcinoembryonic antigen and other glycoconjugates act as ligands for galectin-3 in human colon carcinoma cells. Cancer Res 1995 ; 55 : 2191-9. Ohlsson B, Tranberg KG, Lundstedt C, Ekberg H, Hederstrom E. Detection of hepatic metastases in colorectal cancer : a prospective study of laboratory and imaging methods. Eur I Surg 1993 ; 159 : 275-81. Ohlsson B, Breland U, Ekberg H, Graffner H, Tranberg KG. Follow-up after curative surgery for colorectal carcinoma. Randomized comparison with no follow-up. Dis Colon Rectum 1995 ; 38 : 619-26. Onetto M, Paganuzzi M, Secco GB, Fardelli R, Santi F, Rovida S, et al. Preoperative carcinoembryonic antigen and prognosis in patients with colorectal cancer. Biomed Pharmacother 1985 ; 39 : 392-5. Oshima M, Oshima H, Kobayashi M, Tsutsumi M, Taketa MM. Evidence against dominant negative mechanisms of intestinal polyp formation by APC gene mutations. Cancer Res 1995 ; 55 : 1811-6. Oshima M, Takahashi M, Oshima H, et al. Effects of docohexanoic acid DHA on intestinal polyp development in APC.716 knockout mice. Carcinogenesis 1995 ; 16 : 2605-7. Ovaska J, Jarvinen H, Kujari H, Perttila I, Mecklin JP. Follow-up of patients operated on for colorectal carcinoma. Am I Surg 1990 ; 159 : 593-6. Papkoff J, Rubinfeld B, Schryver B, Polakis P. Wnt-1 regulates free pool of catenins and stabilizes APC-catenin complexes. Mol Cell Biol 1996 ; 16 : 2128-34. Patel PS, Raval GN, Rawal RM, Patel GH, Balar DB, Shah PM, et al. Comparison between serum levels of carcinoembryonic antigen, sialic acid and phosphohexose isomerase in lung cancer. Neoplasma 1995 ; 42 : 271-4. Patt YZ, Podoloff DA, Curley S, Kasi L, Smith R, Bhadkamkar V, et al. Technetium 99m-labeled IMMU-4, a monoclonal antibody against carcinoembryonic antigen, for imaging of occult recurrent colorectal cancer in patients with rising serum carcinoembryonic antigen levels. I Clin Oncol 1994 ; 12 : 489-95. Peethambaram P, Weiss M, Loprinzi CL, Novotny P, O'Fallon JR, Erlichman C, et al. An evaluation of postoperative follow-up tests in colon cancer patients treated for cure. Oncology 1997 ; 54 : 287-92. Peifer M. 1997. beta -catenin as oncogene : the smoking gun. Science 275 : 1752- 1753. Peifer MD, Sweeton D, Casey M, Wieschuas H. Wingless and Zest-white 3 kinase trigger opposing changes in the intracellular distribution of armadillo. Development 1994 ; 120 : 369-80. Piazza GA, Rahm AL, Krutzsch M, et al. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth in inducing apoptosis. Cancer Res 1995 ; 55 : 3110-6. Pietra N, Sarli L, Costi R, Ouchemi C, Grattarola M, Peracchia A. Role of follow-up in management of local recurrences of colorectal cancer : a prospective, randomized study. Dis Colon Rectum 1998 ; 41 : 1127-33. Polakis P. Mutations in the APC gene and their implications for protein structure and function. Curr Opin Genet Dev 1995 ; 5 : 66-71. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992 ; 359 : 235-7. Primrose JN, Bleiberg H, Daniel F, Van Belle S, Mansi JL, Seymour M, et al. Marimastat in recurrent colorectal cancer : exploratory evaluation of biological activity by measurement of carcinoembryonic antigen. Br I Cancer 1999 ; 79 : 509-14. Pugliese V, Aste H, Saccomanno S, Bruzzi P, Bonelli L, Santi L. Outcome of follow-up programs in patients previously resected for colorectal cancer. Tumori 1984 ; 70 : 203-8. Quentmeier A, Moller P, Schwarz V, Abel U, Schlag P. Carcinoembryonic antigen, CA 19-9, and CA 125 in normal and carcinomatous human colorectal tissue. Cancer 1987 ; 60 : 2261-6. Quentmeier A, Schlag P, Hohenberger P, Schwarz V, Abel U. Assessment of serial carcinoembryonic antigen : determinations to monitor the therapeutic progress and prognosis of metastatic liver disease treated by regional chemotherapy. I Surg Oncol 1989 ; 40 : 112-8. Reiter W, Stieber P, Reuter C, Nagel D, Lau-Werner U, Pahl H, et al. Preoperative serum levels of CEA and CA 19-9 and their prognostic significance in colorectal carcinoma. Anticancer Res 1997 ; 17 : 2935-8. Resnik E. 1997. beta -catenin one player two games. Nature Genet 16 : 9-11. T. Reya, H. Clevers, Wnt signalling in stem cells and cancer. Nature 434,. 843-850 (2005). 26. Y. N. Lee, C. C. Malbon, H. Y. Wang, G alpha 13 signals via ... Rittgers RA, Steele G, Zamcheck N, Loewenstein MS, Sugarbaker PH, Mayer RJ, et al. Transient carcinoembryonic antigen (CEA) elevations following resection of colorectal cancer : a limitation in the use of serial CEA levels as an indicator for second-look surgery. I Natl Cancer Inst 1978 ; 61 : 315-8. Rocklin MS, Senagore AJ, Talbott TM. Role of carcinoembryonic antigen and liver function tests in the detection of recurrent colorectal carcinoma. Dis Colon Rectum 1991 ; 34 : 794-7. Rojas M, DeMarte L, Screaton RA, Stanners CP. Radical differences in functions of closely related members of the human carcinoembryonic antigen gene family. Cell Growth Differ 1996 ; 7 : 655-62. Rosin-Arbesfeld R, Townsley F, Bienz M. The APC tumour suppressor has a nuclear export function. Nature 2000 ; 406 : 1009-12. Ross DD, Gao Y, Yang W, Leszyk J, Shively J, Doyle LA. The 95-kilodalton membrane glycoprotein overexpressed in novel multidrug-resistant breast cancer cells is NCA, the nonspecific cross- reacting antigen of carcinoembryonic antigen. Cancer Res 1997 ; 57 : 5460-4. Rubinfeld B., Robbins P., El-Gamil M., Albert I., Porfiri E., Polakis P. 1997. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 275 : 1790-1792. Rubinfield B., et al. 1993. Association of the APC gene product with betacatenin. Science 262: 1731-1734. Rubinfield B., Albert A., Porfiri E., Fiol C., Munemitsu S., Polakis P. 1996. Binding of the GSK3-beta to the APC-beta-catenin complex and regulation of complex assembly. Science 272 : 1023-1025. Rubinfeld B, Albert I, Porfiri E, Munemitsu S, Polakis P. Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res 1997 ; 57 : 4624-30. Ruibal MA. CEA serum levels in non-neoplastic disease. Int .1 Biol Markers 1992 ; 7 : 160-6. Rymer JC, Sabatier R, Daver A, Bourleaud J, Assicot M, Bremond J, et al. A new approach for clinical biological assay comparison and standardization : application of principal component analysis to a multicenter study of twenty-one carcinoembryonic antigen immunoassay kits. Clin Chem 1999 ; 45 : 869-81. Scheele J, Stang R, Altendorf-Hofmann A, Paul M. Resection of colorectal liver metastases. World I Surg 1995 ; 19 : 59B71. Schwartz MK. Cancer markers. In : De Vita VTJ, Hellman S, Rosenberg SA, eds. Cancer principle and practice of oncology. Philadelphia : Lippincott. 1993 : 531B42. Scott NA, Wieand HS, Moertel CG, Cha SS, Beart RW, Lieber MM. Colorectal cancer. Dukes' stage, tumor site, preoperative plasma CEA level, and patient prognosis related to tumor DNA ploidy pattern. Arch Surg 1987 ; 122 : 1375B9. Shahangian S, Fritsche HA, Hughes JI. Carcinoembryonic antigen in serum of patients with colorectal polyps: correlation with histology and smoking status. Clin Chem 1991 ; 37 : 651B5. Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the APC gene. Science 1997 ; 278 : 120B3. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 1999 ; 96 : 5522B7. Slentz K, Senagore A, Hibbert J, Mazier WP, Talbott TM. Can preoperative and postoperative CEA predict survival after colon cancer resection ? Am Surg 1994 ; 60 : 528B31. Smith K.J., Levy D.B., Maupin P., Pollard T.D., Vogelstein B., Kinzler K.W. 1994. Wild type but not mutant APC associates with the microtubule cytoskeleton. Cancer Res 54 : 3672B3675. Smith KJ, Johnson KA, Bryan T, et al. The APC gene product in normal and tumor cells. Proc Natl Acad Sci USA 1993 ; 90 : 2846B50. Smits R, Kielma MF, Breukel C, Zurcher C, Neufeld K, Jagmohan-Changur S, et al. Apc1638T : a mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev 1999 ; 13 : 1309-21. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998 ; 58 : 1130-4. Staab HJ, Anderer FA, Stumpf E, Fischer R. Slope analysis of the postoperative CEA time course and its possible application as an aid in diagnosis of disease progression in gastrointestinal cancer. Am J Surg 1978 ; 136 : 322-7. Staab HJ, Anderer FA, Brummendorf T, Stumpf E, Fischer R. Prognostic value of preoperative serum CEA level compared to clinical staging. I. Colorectal carcinoma. Br J Cancer 1981 ; 44 : 652-62. Staab HJ, Brummendorf T, Hornung A, Anderer FA, Kieninger G. The clinical validity of circulating tumor-associated antigens CEA and CA 19-9 in primary diagnosis and follow-up of patients with gastrointestinal malignancies. Klin Wochenschr 1985 ; 63 : 106-15. Staab HJ, Anderer FA, Stumpf E, Hornung A, Fischer R, Kieninger G. Eighty-four potential second-look operations based on sequential carcinoembryonic antigen determinations and clinical investigations in patients with recurrent gastrointestinal cancer. Am J Surg 1985 ; 149 : 198-204. Staib L, Schirrmeister H, Reske SN, Beger HG. Is (18)F-fluorodeoxyglucose positron emission tomography in recurrent colorectal cancer a contribution to surgical decision making ? Am J Surg 2000 ; 180 : 1-5. Steele G, Ellenberg S, Ramming K, O'Connell M, Moertel C, Lessner H, et al. CEA monitoring among patients in multi-institutional adjuvant G.I. therapy protocols. Ann Surg 1982 ; 196 : 162-9. Stevens DP, Mackay IR, Cullen KJ. Carcinoembryonic antigen in an unselected elderly population : a four year follow up. Br J Cancer 1975 ; 32 : 147-51. Su L.K., Vogelstein B., Kinzler K.W. 1993. Association of the APC tumor suppressor protein with catenins. Science 262 : 1734-1737. Su L.K., et al. 1995. APC binds to the novel protein EB1. Cancer Res 55 : 2972- 2977. Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992 ; 256 : 668-70. Szymendera JJ, Nowacki MP, Kozlowicz-Gudzinska I, Kowalska M. Value of serum levels of carcinoembryonic antigen, CEA, and gastrointestinal cancer antigen, GICA or CA 19-9, for preoperative staging and postoperative monitoring of patients with colorectal carcinoma. Dis Colon Rectum 1985 ; 28 : 895-9. Szymendera JJ, Nowacki MP, Szawlowski AW, Kaminska JA. Predictive value of plasma CEA levels : preoperative prognosis and postoperative monitoring of patients with colorectal carcinoma. Dis Colon Rectum 1982 ; 25 : 46-52. Takahashi T, Kato T, Kodaira S, Koyama Y, Sakabe T, Tominaga T, et al. Prognostic factors of colorectal cancer. Results of multivariate analysis of curative resection cases with or without adjuvant chemotherapy. Am J Clin Oncol 1996 ; 19 : 408-15. Tartter PI, Slater G, Gelernt I, Aufses AH. Screening for liver metastases from colorectal cancer with carcinoembryonic antigen and alkaline phosphatase. Ann Sur 1981 ; 193 : 357-60. Tate H. Plasma CEA in the post-surgical monitoring of colorectal carcinoma. Br J Cancer 1982 ; 46 : 323-30. Thomas G. Dix ans de recherche sur les prédispositions génétiques au développement des tumeurs. meclecine/sciences 1995 ; 11 : 336-48. Thomas G, Muleris M, Salmon RJ. La génétique du cancer colorectal. meclecine/sciences 1988 ; 4 : 274-80. Thomas P, Gangopadhyay A, Steele G, Andrews C, Nakazato H, Oikawa S, et al. The effect of transfection of the CEA gene on the metastatic behavior of the human colorectal cancer cell line MIP-101. Cancer Letters 1995 ; 92 : 59-66. Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family : molecular biology and clinical perspectives. J Clin Lab Anal 1991 ; 5 : 344-66. Thompson J, Seitz M, Chastre E, Ditter M, Aldrian C, Gespach C, et al. Down-regulation of carcinoembryonic antigen family member 2 expression is an early event in colorectal tumorigenesis. Cancer Res 1997 ; 57 : 1776-84. Tibbetts LM, Doremus CM, Tzanakakis GN, Vezeridis MP. Liver metastases with 10 human colon carcinoma cell lines in nude mice and association with carcinoembryonic antigen production. Cancer 1993 ; 71 : 315-21. Tobaruela E, Enriquez JM, Diez M, Camunas J, Muguerza J, Granell J. Evaluation of serum carcinoembryonic antigen monitoring in the follow-up of colorectal cancer patients with metastatic lymph nodes and a normal preoperative serum level. Int J Biol Markers 1997 ; 12 : 18-21. Turner DM, Williams DM, Sankaran D, Lazarus M, Sinnott PJ, Hutchinson IV. Eur J Immunogenet 1997; 24:1-8. ( AN : 97196782 ). Interleukin-10 (IL-10) has been ... Ullmann CD, Schlom J, Greiner JW. Interleukin-6 increases carcinoembryonic antigen and histocompatibility leukocyte antigen expression on the surface of human colorectal carcinoma cells. J Immunother 1992 ; 12 : 231-41. Umehara Y, Kimura T, Yoshida M, Oba N, Harada Y. Comparison of doubling times of serum carcinoembryonic antigen produced by various metastatic lesions in recurrent gastric and colorectal carcinomas. Cancer 1993 ; 71 : 4055-9. Van der Schouw YT, Verbeek AL, Wobbes T, Segers MF, Thomas CM. Comparison of four serum tumour markers in the diagnosis of colorectal carcinoma. Br J Cancer 1992 ; 66 : 148-54. Van de Wetering M., et al. 1997. Armadillo coactivates transcription driven by the product of the drosophila segment polarity gene dTCF. Cell 88 : 789-799. Van Solinge WW, Nielsen FC, Friis-Hansen L, Falkmer UG, Rehfeld JF. Expression but incomplete maturation of progastrin in colorectal carcinoma. Gastroenterology 1993 ; 104 : 1099-107. Verazin G, Riley WM, Gregory J, Tautu C, Prorok JJ, Alhadeff JA. Serum sialic acid and carcinoembryonic levels in the detection and monitoring of colorectal cancer. Dis Colon Rectum 1990 ; 33 : 139-42. Verdi CJ, Ahmann FR, Schifman RB, Elvick AL, Ahmann ME, Marx PC. Comparative evaluation of serum CA 195 and carcinoembryonic antigen in metastatic carcinoma. Cancer 1993 ; 71 : 3625-32. Verhaar MJ, Damen CA, Zonnenberg BA, Blijham GH. In vitro upregulation of carcinoembryonic antigen expression by combinations of cytokines. Cancer Letters 1999 ; 139 : 67-73. Verhelst J, Vanden Broecke E, Van Meerbeeck J, De Backer W, Blockx P, Vermeire P. Calculation of half-life of carcinoembryonic antigen after lung tumour resection : a case report. Eur Respir 1 1991 ; 4 : 374-6. Vogelstein B, Fearon E, Hamilton S, et al. Genetic alterations during colorectal tumor development. N Engl I Med 1988 ; 319 : 525-32. Von Kleist S, Hesse Y, Kananeeh H. Comparative evaluation of four tumor markers, CA 242, CA 19/9, TPA and CEA in carcinomas of the colon. Anticancer Res 1996 ; 16 : 2325-31. Wanebo HJ, Llaneras M, Martin T, Kaiser D. Prospective monitoring trial for carcinoma of colon and rectum after surgical resection. Surg Gynecol Obstet 1989 ; 169 : 479-87. Wang JY, Tang R, Chiang JM. Value of carcinoembryonic antigen in the management of colorectal cancer. Dis Colon Rectum 1994 ; 37 : 272-7. Wang JY, Chiang JM, Jeng LB, Changchien CR, Chen JS, Hsu KC. Resection of liver metastases from colorectal cancer : are there any truly significant clinical prognosticators ? Dis Colon Rectum 1996 ; 39 : 847-51. Wang TC, Koh TJ, Varro A, Cahill RJ, Dangler CA, Fox JG, et al. Processing and proliferative effects of human progastrin in transgenic mice. I Clin Invest 1996 ; 98 : 1918-29. Ward U, Primrose JN, Finan PJ, Perren TJ, Selby P, Purves DA, et al. The use of tumour markers CEA, CA-195 and CA-242 in evaluating the response to chemotherapy in patients with advanced colorectal cancer. Br I Cancer 1993 ; 67 : 1132-5. Webb A, Scott-Mackie P, Cunningham D, Norman A, Andreyev J, O'Brien M, et al. The prognostic value of CEA, beta HCG, AFP, CA125, CA19-9 and C-erb B- 2, beta HCG immunohistochemistry in advanced colorectal cancer. Ann Oncol 1995 ; 6 : 581-7. Wolf RF, Cohen AM. The miniscule benefit of serial carcinoembryonic antigen monitoring after effective curative treatment for primary colorectal cancer. I Am Coll Surgeons 1997 ; 185 : 60-4. Wong MH, Hermiston ML, Syder AJ, Gordon JI. Forced expression of the tumor suppressor adenomatous polyposis coli protein induces disordered cell migration in the intestinal epithelium. Proc Natl Acad Sci USA 1996 ; 93 : 9588-93. Yoshimasu T, Miyoshi S, Maebeya S, Suzuma T, Bessho T, Hirai I, et al. Analysis of the early postoperative serum carcinoembryonic antigen time- course as a prognostic tool for bronchogenic carcinoma. Cancer 1997 ; 79 : 1533-40. Yoshimasu T, Maebeya S, Suzuma T, Bessho T, Tanino H, Arimoto J, et al. Disappearance curves for tumor markers after resection of intrathoracic malignancies. Int .1 Biol Markers 1999 ; 14 : 99-105. Zeng Z, Cohen AM, Urmacher C. Usefulness of carcinoembryonic antigen monitoring despite normal preoperative values in node-positive colon cancer patients. Dis Colon Rectum 1993 ; 36 : 1063-8. Zhang T, Nanney LB, Luongo C, et al. Concurrent expression of cyclin D1 and cyclin-dependent kinase 4 (cdk4) in intestinal adenomas from multiple intestinal neoplasia (Min) mice and human familial adenomatous polyposis patients. Cancer Res 1997 ; 57 : 169-75.
| 
Changeons ce systeme injuste, Soyez votre propre syndic 
"Des chercheurs qui cherchent on en trouve, des chercheurs qui trouvent, on en cherche !" | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||