|
20 novembre 2014
Institut de la Francophonie pour l'Informatique

Mémoire de fin d'études pour l'obtention du
diplôme de Master II Informatique
Option : Réseaux et Systèmes Communicants
Développement d'un portail web pour le
criblage
virtuel sur la grille de calcul
Promotion 17-RSC
Rédigépar :
Louacheni Farida
Sous l'encadrement de :
Dr.Nguyen Hong Quang
Dr.Doan
Trung Tung
Dr.Bui The Quang

Remerciements
Ce travail de stage de fin d'études a
étéeffectuéau sein du Laboratoire MSI à l'Institut
de la Francophonie pour l'Informatique, sous la direction du Docteur
Nguyen Hong Quang, auquel je tiens à exprimer ma
profonde gratitude, et ma vive reconnaissance pour m'avoir confiéce
sujet.
J'adresse mes plus vifs remerciements au Dr.Doan Tung
Tung et Dr.Quang Bui The de m'avoir encadréet
prodiguémaints conseils. Je suis très reconnaissante à
tous les enseignants de l'IFI pour la qualitéde
l'enseignement qu'ils nous ont offerts.
Ma reconnaissance infinie à mes très chers
parents qui m'ont enseignéla persévérance dans mes
études, qui m'ont toujours étéd'un grand secours par leur
soutient et leur encouragement, ainsi mes adorables soeurs et mon très
cher frère et mon ami Yacine-Malek.
Enfin, un immense merci à mes amis qui m'ont toujours
soutenue.

Résumé
À l'heure actuelle, la grille de calcul est en train de
devenir une force motrice majeure pour de nouvelles approches pour la
collaboration de science à grande échelle. Plusieurs programmes
nationaux et internationaux eScience ont favoriséla collaboration entre
chercheurs de différents domaines scientifiques.
Dans le domaine biomédicale, plus précisement
dans la recherche de nouveaux médicaments pour les maladies
infectieuses. La grille de calcul a initiéplusieurs projets à
grande échelle dans les approches de criblage de médicaments
in-silico. Le projet WISDOM a étéparmi les
premiers projets dans le domaine public qui a fait usage de la grille tout en
permettant le docking in-silico pour simuler l'in-teraction de
médicaments potentiels avec des protéines cibles. Le docking
in-silico est la première étape dans le processus de
criblage virtuel, il est considérécomme l'une des approches les
plus prometteuses afin accélérer et de réduire les
coûts de développement de nouveaux médicaments pour les
maladies négligées.
Bien que, de nombreuses applications ont
étédéveloppées pour permettre le criblage virtuel
dont le but d'accélérer le processus de recherche des
médicaments. Une barrière critique de ces programmes est leur
complexitéen terme d'utilisation et de prévoir des
procédures concises pour les utilisateurs réguliers.
L'objectif de ce travail est de développer un portail
web conviviale pour effectuer le criblage virtuel, et de déployer un
très grand nombre de docking sur la grille de calcul. Pour atteindre ce
but, la grille de calcul a étéutilisépour
accélérer la recherche et la découverte de nouveaux
médicaments in-silico et traitements pour les maladies
infectieuses.

Abstract
Grid computing is currently developing into a major driving
force for new approaches towards collaborative large scale science. Several
national and international eScience programs have fostered collaboration
between researchers from different scientific domains.
In the biomedical field, more precisely in drug discovery for
infectious diseases. Grid computing has initiated several projects on large
scale in-silico drug screening approaches. The project WISDOM
was amongst the first projects in the public domain that made use of grid
enabled in-silico docking to simulate the interaction of potential
drugs with target proteins. In-silico docking is the first step in the
virtual screening process, which is one of the most promising approaches to
speed-up and to reduce the costs of the development of new drugs.
Although, many applications have been developed to allow
in-silico screening, but a critical barrier of these programs is the
lack of a suitable, easy, simple way to use and to provide concise procedures
for regular users.
The main goal of this work is to develop a user-friendly web
portal to perform virtual screening and to deploy a large number of docking on
grid computing. To achieve this goal, the grid computing was used to accelerate
research and discovery of new drugs in-silico for infectious
diseases.
i
Table des matières
1 Introduction 1
1.1 Problèmatique 1
1.2 Notre contribution 2
1.3 Plan du mémoire 2
2 État de l'art 3
2.1 Conception de médicaments in-silico 3
2.2 Criblage virtuel »Vitual Screening» 4
2.2.1 Introduction 4
2.2.2 Découverte de nouveaux médicaments avec le
criblage virtuel. . . 6
2.2.3 Les différentes stratégies du criblage
virtuel 6
2.2.4 Criblage virtuel à haut débit 7
2.2.5 Conclusion 7
2.3 Docking 9
2.3.1 Introduction 9
2.3.2 Approches du docking 10
2.3.3 Principe du docking 11
2.3.4 Outils de Docking 12
2.3.5 Conclusion 13
2.4 AutoDock 14
2.4.1 Docking avec AutoDock 15
2.4.2 Conclusion 17
2.5 Grille de calcul 18
2.5.1 Introduction 18
2.5.2 Grille de calcul 18
2.5.3 Organisation virtuelle 19
2.5.4 Architecture générale d'une grille de calcul
20
2.5.5 Composants de la grille 21
2.5.6 Fonctionnement de la grille 22
2.5.7 Avantages & Défis de la grille 24
2.5.8 Conclusion 24
2.6 Portail GVSS 26
2.6.1 Introduction 26
2.6.2 La plate-forme GAP 27
2.6.3 Architecture GVSS 28
2.6.4 Conclusion 29
2.7 Plate-formes utilisés 30
2.7.1 WISDOM 30
2.7.2 DIRAC 33
ii
3 Implémentation 37
3.1 Architecture du système proposée 37
3.2 Outils utilisés 38
3.3 Conception du portail 41
3.4 Développement du portail du web 48
3.4.1 Les services web 48
4 Expérimentation &
Résultats 52
4.1 Conclusion 63
5 Conclusion & perspective
64
iii
Table des figures
|
1
|
Processus de conception de médicaments in-silico
[11]
|
4
|
|
2
|
Criblage Virtuel in-silico
|
5
|
|
3
|
Docking protéine-ligand
|
10
|
|
4
|
Étapes du Docking
|
11
|
|
5
|
Illustration de docking/scoring [6]
|
12
|
|
6
|
Comparaison des programmes de docking [16]
|
13
|
|
7
|
Procédures de docking avec AutoDock
|
15
|
|
8
|
La grille de calcul
|
19
|
|
9
|
Couches de la grille de calcul
|
21
|
|
10
|
Architecture de grille de calcul [10]
|
23
|
|
11
|
Portail GVSS
|
27
|
|
12
|
Architecture Service de criblage virtuel GAP (GVSS) [7]
|
29
|
|
13
|
Architecture WPE [9]
|
31
|
|
14
|
Intergiciel DIRAC
|
33
|
|
15
|
Architecture DIRAC [20]
|
35
|
|
16
|
Architecture du système proposée
|
38
|
|
17
|
Workflow soumission de job sur la grille avec Taverna
|
39
|
|
18
|
Diagramme de classe du portail web
|
42
|
|
19
|
Cas d'utilisation pour le Ligand
|
43
|
|
20
|
Cas d'utilisation pour la Protéine
|
44
|
|
21
|
Cas d'utilisation pour les paramètres de grille
|
45
|
|
22
|
Cas d'utilisation pour le docking
|
46
|
|
23
|
Cas d'utilisation pour l'administrateur du portail
|
47
|
|
24
|
Modèle MVC
|
48
|
|
25
|
Description des services web implémentés
|
49
|
|
26
|
Workflow des services web du portail
|
51
|
|
27
|
Interface d'accueil du portail web
|
52
|
|
28
|
Interface de création d'un nouveau compte
|
53
|
|
29
|
Interface d'authentification
|
53
|
|
30
|
Interface de gestion des utilisateurs
|
54
|
|
31
|
Interface d'ajout d'un nouveau Ligand
|
54
|
|
32
|
Interface de liste des Ligands disponibles
|
55
|
|
33
|
Interface de gestion des protéines
|
55
|
|
34
|
Interface de modification d'une protéine
|
56
|
|
35
|
Interface d'ajout de fichier de paramètres de la grille
|
56
|
|
36
|
Interface d'ajout d'un nouveau projet de docking
|
57
|
|
37
|
Vérification d'ajout du nouveau projet
|
57
|
|
38
|
Soumission de job de docking
|
58
|
|
39
|
Téléchargement du résultat de docking
|
58
|
iv
40
|
Fichier log de docking »dlg»
|
59
|
|
41
|
Soumission du projet de docking ProjectZinc1OKE
|
59
|
|
42
|
Téléchargement du résultat de docking
|
60
|
|
43
|
Enregistrement du résultat du job sur la grille de
calcul
|
60
|
|
44
|
Les fichiers dlg 4 glg du docking
|
60
|
|
45
|
Les fichiers log de docking et de la grille dlg 4 glg
|
61
|
|
46
|
Téléchargement du résultat des jobs
|
62
|
|
47
|
Les fichiers des jobs soumis en parallèle
|
63
|
1
1 Introduction
Par le passé, un grand nombre de médicaments ont
étédécouverts tout simplement grâce à
l'identification de principes actifs extraits de substances naturelles
historiquement utilisées dans la médecine non-conventionnelle, ou
même par hasard, ce qu'on nomme
»sérendipité». Mais plus le nombre de
médicaments connus augmente et plus les probabilités de faire une
telle découverte sont faibles. Par la suite, les avancées dans le
domaine de la synthèse chimique ont conduit à une démarche
de recherche systématique permettant l'élaboration de nouveaux
médicaments de plus grande efficacité. La découverte de
nouveaux médicaments »drug discovery» est un
processus extrêmement long et fastidieux, 12 à 15 ans peuvent
s'écouler entre la découverte de la molécule et la mise
à disposition du médicament auprès des patients. Les
nouvelles méthodes permettant la découverte de nouveaux
médicaments se doivent donc d'innover afin de mettre en évidence
des molécules encore inconnues ayant un certain potentiel
d'activitésur des cibles biologiques connues [Davis et al,2003]. Les
outils mis en place doivent être capables de guider les chimistes
médicinaux dans le choix des molécules à cribler et
à synthétiser.
Les stratégies de criblage virtuel, ou in-silico,
sont donc depuis quelques années employées en tant
qu'alternative ou de façon complémentaire. Ces techniques sont en
général assez faciles à mettre en place, pour un
coût bien moindre que les criblages expérimentaux. De plus,
l'évolution technologique constante de ces dernières
décennies a permis d'accélérer considérablement le
temps de calcul nécessaire à la simulation de systèmes
complexes ou de bases de données de plusieurs milliers de
molécules. Le criblage virtuel est donc aujour-d'hui employédans
de nombreux projets, afin de sélectionner, au sein de vastes librairies
de molécules, un nombre restreint de composés à cribler
expérimentalement.
1.1 Problèmatique
L'axe principal de ce travail se situe dans le domaine de
bio-informatique. Plus précisement dans la recherche et la
découverte de nouveaux médicaments pour les maladies dangereuses
comme: HIV, Ebola, fièvre de dengue,..., par le biais de techniques
informatiques. Le défi se situe au niveau de la conception de nouveaux
médicaments, qui est un processus long et très onéreux, et
au niveau du déployement d'un grand nombre de docking sur la grille de
calcul. Cependant, les outils existants sont en manque de moyen simple pour
fournir des procédures concises pour les utilisateurs réguliers
(biologistes, chimistes, etc) afin d'ar-ranger les ressources pour mener un
amarrage moléculaires massif. Par conséquent, ces derniers
rencontrent plusieurs difficultés et problèmes lors de
l'utilisation de ces applications, ce qui entraîne une grande perte de
temps et d'argent afin d'accélérer la recherche de nouveaux
traitements pour les maladies négligées.
2
1.2 Notre contribution
Notre contribution repose sur le développement d'un
portail web pour le criblage virtuel en utilisant la grille de calcul pour
faciliter la découverte et la recherche de nouveaux médicaments
pour les maladies graves et négligées. Nous proposons une
interface conviviale et facile à utiliser pour les utilisateurs
non-expérimentés (chimistes, biologistes,
médecins....) en informatique et en grille de calcul.
Afin de favoriser l'intéropérabilitéentre le
portail web et les services de grille de calcul, nous proposons une
architecture qui permettra une analyse et un traitement fiable des
requêtes des utilisateurs finaux.
1.3 Plan du mémoire
Ce mémoire sera organiséen 4 parties
présentant respectivement : l'état de l'art,
implémentation & conception, démonstration &
résultats, conclusion & perspectives. Dans la première
partie, un état de l'art est présentéqui passe en revue le
criblage virtuel, le docking, suivie de l'outil AutoDock. Ensuite nous abordons
la technologie de grille de calcul, le portail GVSS et les plate-formes WISDOM
qui est déployée dans la découverte de nouveaux
médicaments et DIRAC. La deuxième partie du mémoire
présente l'implémentation du portail, qui se focalisera sur
l'architecture proposée, la conception et l'implémentation du
portail. L'avant dernière partie porte sur la démonstration du
portail muni des résultats obtenus. À la fin, ce mémoire
ce termine par une conclusion générale et quelques
perspectives.
3
2 État de l'art
Aujourd'hui, les projets scientifiques produisent et analysent
une quantitéd'information sans précédent, ce qui
nécessite une puissance de calcul jamais vue auparavant. Les leaders
dans ce défi de traitement de données sont les expériences
du LHC au CERN, qui accumulent des dizaines de pétaoctets de
donnés chaque année. Cependant, il se révèle que
d'autres domaines scientifiques s'approchent aussi de ces limites. Par
conséquent les utilisateurs devons exploiter les ressources disponibles
à travers le monde de manière aisée et facile. Plusieurs
travaux existent qui illustrent le développement et le
déploiement des applications sur l'infrastructure de la grille de
calcul, et qui ont montréune utilisation efficace des ressources de
cette dernière. Les utilisateurs sont rarement experts en informatique
et en grille de calcul. Pour cette raison ils ont besoin d'un moyen qui
facilite l'accès aux ressources de la grille dont ils ont besoins d'un
coté, et qui cache la complexitéde l'in-frastructure sous-jacente
de l'autre coté. Dans cette partie, nous allons décrire en
détail la technique du criblage virtuel ainsi l'amarrage
»Docking», le principe de la grille de calcul et son
rôle dans la découverte de nouveaux médicaments pour les
maladies négilgées et dangereuses. Puis, nous présentons
le portail GVSS et la plate-forme WISDOM déployer pour accéder
aux services de grille de calcul et DIRAC.
· Nomenclature
- Ligand, une structure,
généralement une petite molécule qui se lie à un
site de liaison.
- Récepteur, une structure,
généralement une protéine qui contient le site de liaison
actif.
- Site de liaison, zones de protéines
actives qui interagissent physiquement avec le ligand pour la formation d'un
composé.
2.1 Conception de médicaments in-silico
La conception de médicaments assistée par
ordinateur emploie la chimie computationnelle pour la découverte,
l'amélioration et l'étude de médicaments et
molécules biologiquement actives. En effet, l'outil informatique aide la
conception de médicaments à des étapes spécifiques
du processus :
· Dans l'identification des composés
potentiellement thérapeutiques, en utilisant le criblage virtuel
»virtual screening».
· Dans le processus d'optimisation de
l'affinitéet de la sélectivitédes molècules
potentielles vers les têtes de série »lead» ou
appelés encore prototypes.
· Dans le processus d'optimisation du lead de
série par rapport aux propriétés pharmacologiques
recherchées tout en maintenant une bonne affinitéde cette
molécule.
4
Toutes ces étapes d'intervention de l'outil
informatique sont présentées dans le schéma
récapitulatif suivant.

FIGURE 1 - Processus de conception de
médicaments in-silico [11]
2.2 Criblage virtuel »Vitual Screening»
L'identification d'une cible pharmaceutique peut se faire par
différentes méthodes. Une fois la cible identifiée
diagnostiquée, il faut tester un ensemble de molécules candidates
sur cette cible, selon un processus qualifiéde screening. On
distingue deux types de criblage : le criblage virtuel, qui est
réaliséin-silico , tout en permettant la
réalisation de manière
rapide et à moindre coût des prédictions
de l'activitédes molécules. Et le criblage réel
àhaut débit, quand à lui il permet de tester
rapidement »in-vitro» l'activitéde composés
biologiques, et cela est limitépar le nombre de
composés à tester en un temps raisonnable et par le coût
des tests.
2.2.1 Introduction
Le terme criblage virtuel ou »Virtual Screening»
regroupe un ensemble de techniques computationnelles ayant pour objectif
l'exploration de bases de composés à la recherche de nouvelles
molécules. Une analogie souvent utilisée compare ces techniques
à des filtres qui permettraient de constituer des ensembles de
molécules partageant certaines propriétés et de
sélectionner les plus susceptibles d'interagir avec une cible
donnée [13].
Aujourd'hui, le criblage virtuel est largement
utilisépour identifier de nouvelles substances bio-active et pour
prédire la liaison d'une grande base de donnée de ligands
à une cible particulière, dans le but d'identifier les
composés les plus prometteurs. Il s'agit d'une méthode qui vise
à identifier les petites molécules pour l'interaction avec les
sites de protéines cibles afin de faire des analyses et des traitements
ultérieures. Plus précisement, le criblage virtuel est
défini comme l'évaluation automatique de très grandes
banques de composés à l'aide de programmes informatiques, il se
référe à une série in-silico, qui est
une
5
technique effectuer à base d'ordinateur ou par
l'intermédiaire des modèles mathématiques et des
simulations informatique, qui aide dans la découverte de nouveaux
médicaments et de déterminer de nouveaux composés les plus
susceptibles pour se lier à une molécule cible d'une structure 3D
connue [2].

FIGURE 2 - Criblage Virtuel
in-silico
(http: //
serimedis. inserm.
fr )
Compte tenu de l'augmentation rapide du nombre de
protéines, le criblage virtuel continue à croitre comme une
méthode efficace pour la découverte de nouveaux inhibiteurs et de
nouveaux médicaments. Il est utilisédans les premières
phases du développement de nouveaux médicaments. Il a pour but de
sélectionner au sein de chimiothèques varièes des
ensembles réduits de molécules dont le potentiel
d'activitéenvers la cible thérapeutique visée est
supérieur à celui des autres molécules [Enyedy Egan,
2008], c-à-d, les molécules qui peuvent influencer
l'activitéde la protéine cible. Dans ce cas, le criblage a pour
objectif l'identification des motifs structuraux essentiels dans la liaison
ligand-récepteur, et la discrimination des meilleurs composés au
sein de chimiothèques orientées comprenant des molécules
appartenant à une même série.
Le criblage virtuel est très utile et
considérécomme un outil efficace pour accélérer la
découverte de nouveaux traitements et la recherche des
bibliothèques de petites molécules afin d'identifier les
structures qui sont les plus susceptibles de se lier à une cible de
médicament, généralement un récepteur de
protéine [14]. Il dépend de la quantitéd'in-formation
disponibles sur la cible d'une maladie particulière. Les techniques de
criblage virtuel sont devenues des outils indispensables dans la chimie
médicinale qui offrent un moyen d'améliorer la phase de
découverte de médicaments. Elles sont utilisées de
manière quotidienne aussi bien dans les laboratoires de recherche
publics que dans les grands laboratoires pharmaceutiques.
6
2.2.2 Découverte de nouveaux médicaments
avec le criblage virtuel
Le criblage virtuel est la stratégie in-silico
la plus utilisée pour l'identification de composés
(»hits») dans le cadre de la recherche de nouveaux
médicaments. Celui-ci fait désormais partie intégrante de
la plupart des programmes de recherche de composés bioactifs, que
ceux-ci se déroulent en milieu académique ou industriel, car il
constitue un complément essentiel au criblage biologique
haut-débit. Le criblage virtuel permet l'exploration de grande
chimiothèques ( > 106 molécules) à la
recherche de composés actifs vis-à-vis d'une cible
thérapeutique donnée. Ce processus vise à réduire
de façon significative la chimiothèque de départ à
une liste limitée de composés jugés les plus prometteurs.
Cette approche conduit souvent à une nette amélioration de la
»concentration» de molécules actives pour la cible
»hit-rate», tandis qu'une sélection aléatoire
de molécules de la chimiothèque ne saurait fournir un tel
enrichissement. Ainsi, le temps aussi bien que les coûts de
l'identification de nouveaux composés peuvent être réduits
de façon remarquable. Plus précisément, le recours au
criblage in-silico, en préalable à un criblage
biologique à plus petite échelle, permet d'ajuster au mieux le
nombre de tests expérimentaux »in-vitro» en fonction
des contraintes budgétaires et temporelles. Quand les conditions le
permettent, le criblage biologique peut être employéen
parallèle au criblage virtuel, afin d'èvaluer
l'efficacitéde ce dernier et de pouvoir améliorer les
paramètres des programmes informatiques utilisés. La pertinence
de la molécule employée est la première condition pour le
succèes d'un criblage virtuel, bien avant celle des algorithmes
utilisés pour la recherche de touches au sein de la molécule. En
effet, seule une librairie de composés suffisamment diverse peut
garantir une exploration satisfaisante de l'espace chimique, maximisant ainsi
les chances de découvrir de nouveaux composés. Par ailleurs, pour
éviter de perdre du temps avec des molécules possédant des
caractéristiques incompatibles avec celles de composés
d'intérêt pharmaceutique, le processus de criblage comporte
généralement une étape préliminaire de filtrage.
Cette tâche, qui peut être prise en charge par des programmes
spécialisés, consiste à exclure les composés
toxiques. Ensuite, ne sont retenus que les composés obéissant
à des définitions empiriques simples du profil de molécule
active.
2.2.3 Les différentes stratégies du
criblage virtuel
Suivant la nature de l'information expérimentale
disponible, on distingue deux approches distinctes pour le criblage virtuel. La
première se base sur la structure de la cible, qui est connue sous le
nom de »structure-based virtual screening», qui rapporte
souvent aux algorithmes de docking protéine-ligand. Elle consiste
à estimer la complémentaritéstruc-turale de chaque
molécule criblée avec le site actif considéré. En
revanche, ces méthodes sont généralement plus
coûteuses en puissance de calcul et leur emploi requiert souvent une
expertise plus importante.
7
La seconde, reposant sur la connaissance d'un nombre suffisant
d'information concernant une ou plusieurs molécules actives de
référence, est appelée »ligand-based virtual
screening». Cette approche est rapide et relativement simple à
mettre en oeuvre, mais son majeur inconvénient est
l'interdépendance envers les informations de référence
utilisées pour construire le modèle de prédiction
d'affinité. Bien que ces deux approches soient surtout utilisées
de manière exclusive, leur combinaison lors du criblage permet de
maximiser les chances de succès pour identifier de nouvelles touches
»hits». Dans le cadre de ce travail, nous utilisons
l'approche »structure-based».
2.2.4 Criblage virtuel à haut débit
La simulation de docking moléculaire est une
procédéutile pour la prédiction des potentiels interaction
des complexes de petite molécule dans des sites de liaison de
protéines, ces informations sont indispensables dans la conception de
médicaments basée sur la structure (SBDD) »Structure
Based Drug Discovery» [4]. Plusieurs programmes de docking, comme
DOCK, GOLD, Autodock, Glide, LigandFit et FlexX, etc se sont montrés
utiles dans le pipeline de la découverte in-silico de
médicaments. La méthode de base derrière la simulation de
docking moléculaire est de générer toutes les
conformations possibles d'une molécule de docking et évaluer
entre eux l'orientation la plus favorable en tant que mode de liaison de la
molécule à l'aide d'une fonction de scoring. Une
recherche exhaustive sur toutes les conformations correctes d'un
composéest un processus qui consomme beaucoup de temps. Par
conséquent, une simulation de docking efficace pour le criblage à
grande échelle à haut débit (HTS) consommera de grandes
ressources informatiques. Il nécessite quelques Tera-flops par
tâche pour effectuer le docking de milliers de composés pour une
protéine cible. Cependant, les outils existants manquent de moyen simple
pour prévoir des procédures de façon concise pour les
utilisateurs régulier afin d'organi-ser les ressources pour mener un
amarrage moléculaires massives. La technologie de la grille commence une
nouvelle ère de criblage virtuel en raison de son efficacitéainsi
que son rapport coût-efficacité. Le coût des tests
in-vitro traditionnelle est généralement très
élevélors du criblage à grande échelle est
menée. Le criblage virtuel fournit aux scientifiques un outil efficace
pour sélectionner les potentiels composés pour les tests
in-vitro. En conséquence, le criblage virtuel à haut
débit pourrait bien économiser énorme somme d'argent
comparant aux tests in-vitro classique.
2.2.5 Conclusion
Nous avons introduit le concept, les stratégies de
criblage virtuel. Ce dernier est une approche informatique visant à
prédire des propriétés de librairies de molécules.
Avec l'essor considérable de données expérimentales
publiquement disponibles, cette discipline a enregistrédes
progrès considérables quant au débit, la qualitéet
la diversitédes prédictions possibles. Un inventaire des
applications du criblage »in-silico» est donné, tout
en gardant
8
une attention particulière à des cas concrets
d'utilisation ainsi qu'au développements futur. Le criblage virtuel
fournit une solution complémentaire pour le criblage virtuel à
haut débit »HTS», oùil comprend des techniques
de calcul novatrices. L'avantage du criblage virtuel »in-silico»
est donc de fournir une petite liste de molécules à tester
expérimentalement et ainsi réduire les coûts et gagner du
temps. On peut aussi explorer rapidement de nombreuses molécules pour se
focaliser ensuite, au niveau expérimental, sur les molécules les
plus intéressantes. Les difficultés intrinsèques aux
techniques à haut débit ainsi que celles rencontrées lors
des étapes d'optimisation des molécules chimiques, ont
encouragéle développement de nouvelles approches, telles que les
techniques de criblage virtuel par docking moléculaire.
9
2.3 Docking
2.3.1 Introduction
La modélisation de la structure d'un complexe
protéine-ligand est très importante pour la compréhension
des interactions de liaison entre un composépotentiel
»ligand» et sa cible thérapeutique
»protéine», et pour la conception de
médicaments à base de structure moderne.
Le docking ou »amarrage, arrimage» est une
procédétrès utile qui vise à prédire
l'inter-action potentiel de la structure d'un complexe moléculaire
à partir des petites molécules dans les sites de liaison de
protéine afin d'accélérer la recherche et la
découverte de nouveaux médicaments in-silico
(c-à-d à l'aide d'ordinateur), »Le docking
in-silico est la détermination de la structure 3D des complexes
protéiques à l'échelle atomique, qui permet de mieux
comprendre la fonction biologique de ces complexe [1]». Plus
précisement, le docking consiste à trouver la meilleure position
d'un ligand (petite molécule) dans le site de liaison d'un
récepteur (protéine) de façon à optimiser les
interactions avec un récepteur, évaluer les interactions
ligand-protéine de façon à pouvoir discriminer entre les
positionnement observées expérimentalement et les autres. De
façon générale, le docking a pour but de simuler
l'interaction entre les molécules in-silico, et les
résultats obtenus servent à prédire la structure et les
propriétés de nouveaux complexes [3].
Historiquement, les premiers outils de docking
obéissaient au principe dit: »lock-and-key» (principe
clef-serrure), selon lequel le ligand qui représente la clef, est
complémentaire au niveau géométrique du site actif du
récepteur, qui représente la serrure [Yuriev et al., 2011]. Les
ligands sont des petites molécules destinées à inhiber
l'activitéd'une protéine, qui constitue le récepteur. Il
permet aussi, de prédire la structure intermoléculaire entre deux
molécules en une structure tridimensionnelle 3D, les modes de liaison ou
les conformations possibles d'un ligand à un récepteur, et de
calculer l'énergie de liaison. La technique de docking prévoit
également la résistance de la liaison, l'énergie du
complexe, les types de signaux produits et estime l'affinitéde liaison
entre deux molécules. Elle joue un rôle très important dans
l'aide à la décision, afin de déterminer quel ligand
candidat interagira le mieux avec un récepteur protéine cible
[15].
Le docking protéine-ligand est utilisépour
vérifier la structure, la position et l'orientation d'une
protéine quand elle interagit avec les petites molécules comme
les ligands. Son but
est de prédire et de classer les structures
résultant de l'association entre un ligand donnéet une
protéine cible d'une structure 3D connue.

10
FIGURE 3 - Docking protéine-ligand
Le docking ligand-protéine reste donc la méthode
la plus souvent employée, car elle permet une évaluation rapide
de bases de milliers, voire de millions de molécules.
En principe, un programme de docking doit être capable
de générer les modes de liaison attendus pour des ligands dont la
position adoptée au sein du site actif est connue dans un temps
raisonnable. Pour cela, il est nécessaire que l'algorithme de recherche
confor-mationnelle puisse explorer l'espace conformationnel le plus
exhaustivement possible et de façon efficace. Classiquement, on juge la
qualitédu docking en mesurant le RMSD (Root Mean Square Deviation)
sur les atomes entre la pose obtenue en docking, et la pose
observée expérimentalement si elle existe.
2.3.2 Approches du docking
Les différentes approches du docking se distinguent au
niveau de leurs conditions d'ap-plication et de la nature des informations
qu'elles peuvent fournir. La pertinence du choix d'un programme de docking
donnérepose en premier lieu sur l'adéquation entre ces
caractéristiques et celles du système étudié.
L'efficacitéde l'algorithme choisi sera par ailleurs un compromis entre
la rapiditéd'exécution et la précision des
résultats.
Aussi en fonction du but recherchéet du besoin de
précision voulu, trois degrés sont en général
considérés : rigide (les molécules sont
considérées comme rigides), semi-flexible (une molécule
rigide et l'autre flexible), flexible (les deux flexibles). Le niveau
semi-flexible est souvent appliquédans le cas protéine-ligand
oùune des deux molécules (le ligand) de taille moindre est
considérée comme flexible et la protéine comme rigide de
façon à ne pas trop complexifier le système.
11
Le processus de docking consiste à faire interagir une
petite molécule organique avec le récepteur,
généralement de nature protéique. La technique de docking
comprend 4 étapes principales :
1. Préparer les fichiers pour la protéine.
2. Préparer les fichiers pour le ligand.
3. Préparer les fichiers de paramètres pour la
grille.
4. Préparer les fichiers de paramètres pour le
docking. Le schéma ci-après montre clairement les étapes
de docking.

FIGURE 4 - Étapes du Docking
2.3.3 Principe du docking
Le docking moléculaire s'accomplit en deux
étapes complémentaires. La première est le Docking,
qui consiste à rechercher les conformations du ligand capables
à établir des interactions idéales avec le
récepteur en utilisant des algorithmes de recherche: algorithme
génétique, la méthode de Monte Carlo (qui utilise des
procédés aléatoires)... La deuxième dite le
»Scoring», qui sont des méthodes mathématiques
et des fonctions discriminant les poses de docking correctes de celles
incorrectes. Ces méthodes sont utilisées pour estimer la
puissance d'interaction et l'affinitéde liaison et qui permet
d'évaluer les conformations par un calcul rapide d'énergie
d'interaction des ligands avec un récepteur pour ne retenir que la
meilleure.
12
La formule utilisée pour le scoring est la suivante :
AG= Acomplexe - Aligand
- Aprotéine
La figure ci-dessous schématise le principe du
docking/scoring, oùR symbolise une structure du
récepteur. Tandis que, A, B et C représentent les petites
molécules.

FIGURE 5 - Illustration de docking/scoring
[6]
Le docking peut être interprétéde
manière qualitative par observation de l'entitéligand
dans la cavitéde la protéine, mais également de
manière quantitative par traitement des données provenant des
fonctions de scoring.
2.3.4 Outils de Docking
A l'heure actuelle, plus de 30 programmes de docking
moléculaires (commerciaux ou non) sont disponibles [6]. Les plus
fréquemment cités sont respectivement : AutoDock [9], GOLD,
FlexX, DOCK et ICM. Ils permettent notamment un criblage rapide de vastes
librairies de composés. Ces programmes reposent le plus souvent sur des
algo-
rithmes spécifiques (Algorithme
génétique, Recuit Simulé...), leur protocole est
composéde 2 étapes essentielles Docking/Scoring.
Pour accomplir la tâche de docking, les outils
d'amarrage moléculaire vont générer une série de
poses différentes de liaison au ligand et en utilisant une fonction de
notation »scoring» pour évaluer les affinités
de liaison de ligand pour les poses générées afin de
déterminer le meilleur mode de liaison.

13
FIGURE 6 - Comparaison des programmes de
docking [16]
Comme la figure ci-dessus montre, le programme AutoDock est le
plus citéet le plus utiliséparmi les autres programmes de
docking.
2.3.5 Conclusion
Le processus de docking est l'un des premières
étapes dans la conception de médicaments, il consiste à
faire interagir une petite molécule organique avec un récepteur,
généralement de nature protéique. En conséquent, le
plus grand avantage des méthodes de docking protéine-ligand est
qu'ils peuvent proposer des hypothèses structurelles sur la façon
dont une petite molécule peut interagir avec sa cible
macromolécule. Des études ont montréque certains
algorithmes de docking sont plus fiables que d'autres pour reproduire le mode
de fixation expérimentale de ligand. La contrepartie de ces techniques
est généralement une hausse des temps de calcul et des
ressources. A l'inverse, un projet impliquant le criblage virtuel de millions
de produits ne pourra pas être accompli avec ce type d'algorithme mais
plutôt des codes plus simples, dans lesquels les approximations
engendrent un gain de temps de calcul et d'argent. Le nombre de programme de
docking actuellement disponibles est élevéet n'a
cesséd'augmenter au cours des dernières décennies. Les
exemples suivants présentent un aperçu des programmes les plus
communs de docking protéine-ligand (LigandFit, FlexX, AutoDock). Dans ce
travail nous avons utiliséle programme AutoDock.
Le docking est un type d'application facilement distribuable
sur une grille. De sorte que, de nombreuses ressources de calcul et de stockage
ont étémises à disposition par le projet EGEE
(Enabling Grids for E-sciencE), qui est financépar la
commission européenne et qui a pour but de construire sur les plus
récentes avancées des technologies de grille et de
développer un service d'infrastructure de grille disponible 24h/24h.
14
2.4 AutoDock
AutoDock [4] est un programme flexible, utiliser pour le
docking protéine-ligand. Il s'agit d'un ensemble de procédures,
dont le but de prédire l'interaction de petites molécules, telles
que des médicaments candidats »ligand» ou des
substrats à un récepteur dont la structure 3D est connue.
AutoDock fonctionne essentiellement comme une procédure en deux
étapes : le calcul de la carte d'interaction du site de liaison du
récepteur qui est réaliséavec autogrid,
et la position de ligand sur la carte d'interaction, qui est
effectuée avec autodock.
Le programme AutoGrid est chargéde
calculer les cartes d'interaction des grilles afin de maximiser l'étape
d'évaluation des différentes configurations du ligand. Pour cela
une grille entoure la protéine réceptrice et un atome sonde est
placéà chaque intersection. L'énergie d'interaction de cet
atome avec la protéine est calculée et attribuée à
l'empla-cement de l'atome sonde sur la grille. Une grille d'affinitéest
calculépour chaque type d'atome du ligand. Le temps de calcul de
l'énergie en utilisant les grilles est proportionnel au nombre d'atomes
du ligand uniquement, il est indépendant du nombre d'atomes du
récepteur.
Le programme AutoDock effectue la partie de
recherche et d'évaluation des différentes configurations du
ligand. Il est possible d'utiliser plusieurs techniques pour obtenir les
configurations (par recuit simulé, algorithme génétique ou
par algorithme génétique La-marckien). Pour la méthode
Monte Carlo, à chaque pas un déplacement au hasard de tous les
degrés de libertéest effectué(translation, rotation,
torsion). Les énergies de la nouvelle et de l'ancienne configuration
sont comparées. Si la nouvelle est plus basse elle est gardée,
sinon elle est conservée ou rejetée.
La version actuelle du programme est la version 4.2, qui
fournit de nouvelles fonctionnalités importantes pour le docking comme
la flexibilitédes résidus de protéines et des fonctions de
score de haute qualité. Afin de pouvoir réaliser le criblage
virtuel avec AutoDock, un ensemble d'outils nécessaires doivent
être mis en place :
· Java OpenJDK (openjdk-7-jdk)
· Python 2.7
· AutoDock4.2 (http: //
autodock. scripps.
edu )
· MGLTools (http: // mgltools. scripps. edu/ )
· autodocksuite-4.2.5.1-i86Linux2.tar.gz
· AutoDockTools
· Les fichiers nécessaires
téléchargeable depuis le site de base de donnée de
protéine :
www.pdb.com
15
2.4.1 Docking avec AutoDock
AutoDock a besoin de connaître les types, les charges et la
liste de liaison de chaque atome, afin de pouvoir effectuer la procédure
de docking. Tout d'abord, il faut chercher dans la base de donnée PDB
(Protein Data Bank) dans le site (http: //
www. pdb. org,http: //
www. rcsb. org ),
les fichiers pdb pour la protéine et le ligand.

FIGURE 7 - Procédures de docking avec AutoDock
La procédure de docking avec AutoDock se décompose
en plusieurs étapes :
1. Préparer le fichier d'entrée de
protéine. Dans cette étape un fichier PDBQT(Protein Data
Bank, Partial Charge (Q), & Atom Type (T)) sera créé,
qui contient les atomes et les charges partielles.
> input protein.pdb
> output protein.pdbqt
L'utilisateur possède 2 choix pour préparer son
protéine, soit il utilise l'outil »ADT», soit via la
commande suivante :
> /usr/local/MGLTools-1.5.6/bin/pythonsh
/usr/local/MGLTools-1.5.6 /MGLToolsPckgs/AutoDockTools/Utilities24/prepare
receptor4.py
-r protein.pdb
2.
16
Préparer le fichier d'entrée de ligand. Cette
étape est très semblable à la préparation du
protéine. Nous créons un fichier dont l'extension est PDBQT du
ligand.La préparation s'effectue comme suit :
> input ligand.pdb
> output ligand.pdbqt
> /usr/local/MGLTools-1.5.6/bin/pythonsh
/usr/local/MGLTools-1.5.6
/MGLToolsPckgs/AutoDockTools/Utilities24/
prepare
ligand4.py
-r ligand.pdb
3. Génération d'un fichier de paramètre
de la grille. Maintenant, nous devons définir l'espace en 3D,
qu'AutoDock considèrera pour le docking. Dans cette phase, nous allons
créer les fichier d'entrées pour »AutoGrid4»,
qui permettra de créer les différents fichiers de carte
»map file» et le fichier de données de la grille
»gpf»(grid parameter file).
> input ligand.pdbqt & protein.pdbqt > output
protein.gpf
> /usr/local/MGLTools-1.5.6/bin/pythonsh
/usr/local/MGLTools-1.5.6/MGLToolsPckgs/AutoDockTools/Utilities24/
prepare
gpf4.py
-l ligand.pdbqt -r protein.pdbqt
4. Génération des fichiers de cartes et de
données de la grille. Dans l'étape précédente, nous
avons crééle fichier de paramètres de la grille, et
maintenant nous allons utiliser »AutoGrid4» pour
générer les différents fichiers de cartes et le fichier
principal de données de la grille.
> input protein.pdbqt & protein.gpf >
autogrid4 -p protein.gpf
Après avoir lancéautogrid, plusieurs nouveaux
fichiers avec l'extension map se créent, qui correspondent à
chaque type d'atome de ligand et des fichiers auxiliaires. Ces fichiers sont
importants dans le processus de docking.
5. Génération du fichier de paramètre de
docking. Cette étape consiste à préparer les fichiers de
docking (dpf).
> input ligand.pdbqt & protein.pdbqt > output ligand
protein.dpf
> /usr/local/MGLTools-1.5.6/bin/pythonsh
/usr/local/ MGLTools-1.5.6/MGLToolsPckgs/ AutoDockTools/ Utilities24/prepare
dpf4.py
-l ligand.pdbqt -r protein.pdbqt
17
On peut préparer les fichiers de paramètres pour
la grille et pour le docking sans utiliser l'outil ADT, en utilisant un script
shell (voir annexe) pour préparer ces fichiers. Le
résultat de ce script sont respectivement les fichiers : dpf
»docking parameter file» et gpf »grid parameter
file».
6. À ce stade, nous aurions créétout un
tas de différents fichiers. Cette avant dernière étape
consiste à exécuter autodock avec la commande ci-après
:
> input protein ligand.gpf
> output result.dlg protein ligand.gpf
> autodock4 -p protein ligand.dpf -l
result.dlg
7. La dernière étape sera consacrée
à l'analyse des résultats de docking. Après avoir
terminéavec succès la procédure de docking. Le meilleur
résultat pour le docking, sont les conformations qui possèdent
une basse énergie. AutoDock peut faire une première analyse des
résultats en regroupant les solutions en classes (clusters) en fonction
de leur proximitéspatiale. La mesure de la proximitéentre deux
solutions est calculée par la racine de la moyenne des carrés des
écarts (Root Mean Square Deviation RMSD) de leurs
coordonnées atomiques. Si le RMSD entre molécules est
inférieur à une distance seuil, ces deux solutions sont dans la
même classe. Le seuil de distance est
appelé»tolérance de classe» et sa valeur par
défaut, pour AutoDock, est de 0,5. Ce paramètre est transmis
à AutoDock par le fichier de paramètrage »dpf»
avant le lancement du docking.
2.4.2 Conclusion
Comme nous avons mentionnédans la partie de docking,
l'amarrage avec AutoDock est une procédéqui comporte plusieurs
étapes. Ce qui nécessitera une préparation
préalable des fichiers pour le docking. Le processus de docking est un
sujet essentiel pour progrésser dans la compréhension des
mécanismes d'interaction moléculaires et pour le
développement
d'outils prédictifs dans le domaine de la médecine.
Dans cette partie, nous avons présentéla procédure du
docking avec AutoDock4.2 en utilisant l'outil AutoDockTools, et
nous
avons appliquéles étapes de docking sur un
exemple concret dans le but de comprendre cette technique. qui va nous aider
dans la prochaine phase de lancement des jobs sur la grille de calcul pour
faire le docking.
18
2.5 Grille de calcul
2.5.1 Introduction
Les chercheurs travaillent sur la compréhension des
changements climatiques, les études océanographiques, la
surveillance et la modélisation de la pollution environnementale, la
science des matériaux, l'étude des procédés de
combustion, la conception de médicaments, la simulation des
molécules et le traitement de données dans le domaine de la
physique des particules. Ils ont étéconfrontés à
plusieurs problèmes informatiques, oùils avaient besoin de
processeurs plus puissants, de plus grandes capacités de stockage des
données, de meilleurs moyens d'analyse et de visualisation. Les
récents progrès de la technologie des réseaux très
haut débit courtes et longues distances ont rendu possible la
construction de systèmes répartis de hautes performances,
distribués à l'échelle planétaire dont certains des
constituants sont des grappes de PC ou des calculateurs parallèles.
Cependant les applications scientifiques parallèles sont par nature
gourmandes en ressources de calcul. Il peut être intéressant de
chercher à les exécuter dans le cas o'u les ressources locales,
cluster de laboratoire, centre de calcul ne suffisent plus. Néanmoins,
les ordinateurs d'une entreprise ne travaillent presque jamais à pleine
charge. Exploiter chaque seconde de latence permet de dégager de la
puissance de calcul, ainsi que des espaces de stockage considérables, le
tout pour un coût souvent inférieur à celui d'un
investissement pour l'acquisition d'un nouveau matériel. Les
technologies de grille de calcul ou »Grid Computing»,
permettent de mettre en partage, de façon sécurisée,
les données et les programmes de multiples ordinateurs, qu'ils soient de
bureau, personnels ou super-calculateurs. Ces ressources sont mises en
réseau et partagées grâce à des solutions logicielle
dédiées [5]. Elles peuvent ainsi générer, à
un instant donné, un système virtuel dotéd'une puissance
gigantesque de calcul et une capacitéde stockage en rapport pour mener
à bien des projets scientifiques ou techniques requérant une
grande quantitéde cycles de traitement ou l'accès à de
gros volumes de données.
2.5.2 Grille de calcul
La grille de calcul ou »grid computing» est
une technologie en pleine expansion dont le but d'offrir à la
communautéscientifique des ressources informatiques virtuellement
illimitées. Dans sa version la plus ambitieuse, la grille est une
infrastructure logicielle permettant de fédérer un grand nombre
de ressources de calcul, de bases de données et d'applications
spécialisées distribuées à travers le monde.
Prabhu définit la grille de calcul comme : »Un
ensemble de ressources de calcul distribuésur un réseau local ou
étendu qui apparaît à un utilisateur final ou une large
application en tant que système informatique virtuel» [5]. La
grille de calcul a pour but de réaliser le partage flexible et
coordonner de ressources ainsi que la résolution coopérative de
problème au sein d'organisation virtuelles (VO).
À l'origine, la grille était conçue comme
un grand nombre d'ordinateurs en réseau, oùles
ressources de calcul et de stockage étaient partagées en fonction
des besoins et à la
19
demande des utilisateurs. La grille fournit les protocoles,
les applications et les outils de développement pour réaliser ce
partage dynamiquement et à grande échelle. Ce partage est
hautement contrôlépour définir qui partage quoi, qui
utilise quoi, et sous quelles conditions. Un système de grille est
obligatoirement hautement dynamique puisque les fournisseurs et les
utilisateurs de ressources varient en fonction du temps. Elle permet ainsi de
construire une organisation virtuelle à partir de compétences et
de ressources complémentaires, réparties dans plusieurs
institutions, mais qui seront visibles comme un tout cohérent par les
personnes partageant un objectif commun trop complexe pour être
abordépar une seule équipe. Les technologies de grille permettent
le partage, l'échange, la découverte, la sélection et
l'agrégation de larges ressources hétérogènes,
géographiquement distribués via Internet tels que des capteurs,
des ordinateurs, des bases de données, des dispositifs de visualisation
et des instruments scientifiques. La grille de calcul est largement
utilisédans plusieurs domaines : chimie, bio-informatique,
mathématique, biomédecine...

FIGURE 8 - La grille de calcul 2.5.3
Organisation virtuelle
La grille de calcul prend en charge plusieurs organisations
virtuelles, qui partagent des ressources entre elles. Une Organisation
Virtuelle (VO), est un groupe de chercheurs ayant des intérêts
scientifiques et des exigences scientifiques similaires, qui travaillent en
collaboration avec autres membres et qui partagent des ressources
(données, logiciel, programmes, CPU, espace de stockage),
indépendamment de leur emplacement géographique. Oùchaque
organisation virtuelle gère sa propre liste de membres, selon les
besoins et les objectifs de la VO. Les chercheurs doivent adhérer
à une VO afin d'utiliser les ressources informatiques de la grille
fournie par EGI (https: //
www. egi.
eu ).
20
EGI (European Grid Infrastructure) est une suite du
projet EGEE, qui vise à pérenniser l'infrastructure de grille en
l'ouvrant à toutes les disciplines scientifiques tout en
intégrant les innovations sur le calcul distribué[24]. EGI offre
un support, des services et des outils pour permettre les membres de VO de
profiter de leurs ressources. EGI accueille actuellement plus de 200 VO pour
les communautés ayant des intérêts aussi divers que
sciences de la terre, médecine, bio-informatique, sciences informatiques
et mathématiques ou sciences de la vie.
2.5.4 Architecture générale d'une grille
de calcul
L'architecture d'une grille de calcul est organisée en
couches. Bien que chaque projet ait sa propre architecture, une architecture
générale est importante pour expliquer certains concepts
fondamentaux des grilles, présentés ci-dessous :
· La couche Fabrique (Fabric layer)
C'est la couche de plus bas niveau, elle est en relation
directe avec le matériel afin de mettre à disposition les
ressources partagées. Les ressources fournies par cette couche sont d'un
point de vue physique des ressources telles que des processeurs pour le calcul,
des bases de données, des annuaires ou des ressources réseau.
· La couche réseau (Network
layer)
Elle implémente les principaux protocoles de
communication et d'authentification nécessaire aux transactions sur un
réseau de type grille. Les protocoles de communication permettent
l'échange des données à travers les ressources du niveau
fabrique. Ces protocoles d'authentification s'appuient sur les services de
communication pour fournir des mécanismes sécurisés de
vérification de l'identitédes utilisateurs et des ressources.
· La couche ressource (Resource layer)
Cette couche utilise les services des couches
connectivitéet fabrique pour collecter des informations sur les
caractéristiques des ressources, les surveiller et les contrôler.
La couche ressource ne se préoccupe pas des ressources d'un point de vue
global, elle ne s'intéresse pas à leur interaction, ceci incombe
à la couche collective. Elle ne s'intéresse qu'aux
caractéristiques essentielles des ressources et à la façon
dont elles se comportent.
· La couche collective (Collective
layer)
Elle se charge des interactions entre les ressources. Elle
gère l'ordonnancement et
la co-allocation des ressources en cas de demande des
utilisateurs faisant appel àplusieurs ressources
simultanément. C'est elle qui choisit sur quelle ressource de
calcul faire exécuter un traitement en fonction des
coûts estimés. Elle s'occupe également des services de
réplication des données. En outre, elle est en charge de la
surveillance des services et elle doit assumer la détection des
pannes.
· 21
La couche application (Application layer)
C'est la couche la plus haute du modèle, elle
correspond aux logiciels qui utilisent la grille pour fournir aux utilisateurs
ce dont ils ont besoin, qu'il s'agisse de calcul, ou de données. Les
applications utilisent des services de chacune des couches de
l'architecture.

FIGURE 9 - Couches de la grille de calcul
2.5.5 Composants de la grille
Les principaux composants de l'environnement informatique de
la grille sont discutés en détail dans cette section. Selon la
conception de l'application de la grille et son utilisation prévue,
certains de ces composants mentionnés ci-dessous peuvent ou peuvent ne
pas être nécessaire, et dans certains cas, ils peuvent être
combinés. Les composants de l'infrastructure de la grille de calcul sont
:
· Le portail de la grille
Un portail de grille fournit l'interface pour le service
demandeur (comme les secteurs privé, public et utilisateur commercial),
pour concevoir et accéder à un grand choix de ressources, des
services, des applications et des outils, en encapsulant de la
complexitéde la conception réelle de l'architecture de
réseau sous-jacent à des utilisateurs finaux.
· Service d'information
Le composant de service d'information fournit des
informations sur les ressources disponibles, leur capacités totale, leur
disponibilité, l'utilisation actuelle et les informations de
tarification,... Plus tard cette information est utilisée par le portail
de la grille et le planificateur des ressources pour trouver les ressources
appropriée sur la grille de calcul pour répondre à la
demande de l'utilisateur.
· 22
Courtier de ressources »Resource
Broker»
Le Courtier de ressources ou Resource Broker agit
comme un intergiciel entre le service demandeur (job soumis pour
l'exécution) et un fournisseur de services (ressources disponibles sur
la grille). La tâche d'un courtier de ressources de la grille est
d'identifier dynamiquement les ressources disponibles, pour sélectionner
et allouer les ressources les plus appropriées pour un job
donné.
· Ordonnanceur de ressource
Une fois les ressources ont
étéidentifiées, l'étape suivante consiste à
planifier les travaux en allouant les ressources disponibles. L'ordonnanceur de
ressource doit être utilisé, parce que certains jobs sont
prioritaires par rapport aux autres et certains jobs exigent une longue
autonomie.
· Utilisateur de grille
L'utilisateur de la grille est un consommateur de ressources
de la grille de calcul. Il existe de nombreuses catégories
d'utilisateurs de grille à savoir Les scientifiques, les militaires, les
enseignants et les éducateurs, les entreprises, médecins... Les
catégories d'utilisateurs dépends essentiellement du type de
problème qu'ils vont résoudre sur l'infrastructure du grille.
· Gestionnaire de ressource
Le gestionnaire de ressources de la grille estime les besoins
en ressources, exécute les jobs, contrôle leur état et
retourne les sorties lorsque les jobs sont terminés. Le gestionnaire de
ressources peut consulter le courtier de ressources sur l'affectation des
ressources et assigner les tàaches aux ressources appropriées. En
outre, il doit authentifier l'utilisateur et vérifier s'il est
autoriséà accéder aux ressources avant d'attribuer le
job.
2.5.6 Fonctionnement de la grille
La grille de calcul fonctionne sur le principe de mise en
commun des ressources, oùun grand nombre de ressources de calcul
distribuésont connectées via le réseau à grande
vitesse, et qui sont tous provisionnées en provenant des divers endroits
géographiques et à travers les frontières
organisationnelle. Le fonctionnement de la grille est assez simple. Chaque job
crééest associéà un »jobstep» et
un ensemble de »workunits». Ces unités de travail
sont prêtes à être lancées sur les ressources de la
grille, elles contiennent les informations sur les données, les
paramètres nécessaires ainsi que le programme à
exécuter. Les agents installés sur chaque machine de la grille se
connectent à un intervalle de temps régulier au serveur de grille
pour prendre le job (principe du modèle »pull»).
Avant de télécharger les données, l'agent vérifie
si elles ne sont pas déjàdans son cache, afin d'éviter des
transferts inutiles. L'agent lance alors le programme scientifique. A la
terminaison du programme, l'agent archive les résultats et renvoie
l'archive du résultat au serveur de grille.
23
À chaque job terminéest donc
associéà un ou plusieurs résultats. L'utilisateur
télécharge l'ensemble des résultats. Les étapes
clés pour le fonctionnement du réseau informatique et
l'interaction entre les différents éléments du
réseau sont présentés dans la figure ci-dessous:

FIGURE 10 - Architecture de grille de calcul
[10]
Comme le montre la figure ci-dessus, le fonctionnement des
différents composants de la grille sont :
· Les utilisateurs du réseau présentent leurs
jobs au Resource Broker de la grille.
· Le courtier de ressources »Resource
Broker» de la grille procède à la découverte des
ressources et de la tarification des informations en utilisant le service de
l'informa-tion.
· Le gestionnaire de ressources de la grille
»Resource Manager», authentifie et assure le crédit
nécessaire dans le compte de l'utilisateur afin de déployer les
ressources de la grille.
· L'ordonnanceur de ressource (Resource Scheduler),
exécute alors le job sur les résultats en matière de
ressources et de rendement approprié.
· Le courtier rassemble les résultats et les passent
à l'utilisateur de la grille.
24
2.5.7 Avantages & Défis de la grille
Les avantages d'utiliser une telle architecture sont multiples
et indéniables. Nous pouvons citer les exemples suivants :
· Déploiement des ressources
inutilisées
La grille est un concept au fort potentiel, dont
l'idée est de faire en sorte que toute la puissance de calcul des PCs
inutilisés soit utilisée. De nos jours, les ordinateurs restent
souvent inutilisés pendant de longues périodes, leur processeur
n'étant que rarement utiliséà 100%. Avec cette
technologie, les moments d'inactivitéde cen-
taines ou de milliers d'ordinateurs et de serveurs peuvent
être utilisés et vendus àquiconque ayant besoin
d'une puissance de calculs massive.
· Basésur une architecture de type
client/serveur
La grille de calcul repose sur une architecture bien
précise et très sûre, en l'occur-
rence, c'est l'architecture client/serveur qui a
étéchoisie. Cette architecture a
étéadaptéen fonction des besoins
spécifiques de la technologie de grille de calcul.
· Meilleure rentabilisation du
matériel
Il est évident qu'il y a une sous utilisation des
machines, et la grille présente la solution idéale, d'un point de
vue économique pour les entreprises et d'un point de vue pratique pour
les utilisateurs, pour rentabiliser les ressources.
Les défis de la recherche rencontrés par les
technologies de grilles de calcul actuelles sont répertoriés
comme :
· Dynamicité: Les ressources
dans la grille sont gérées et contrôlées par plus
d'une organisation, en raison de ce que les ressources peuvent rejoindre ou
sous forme de sortie de grille à tout moment, ce qui peut conduire
à plus de charge sur la grille.
· Administration : La technologie de
grille est essentiellement un groupe de ressources mises en commun qui
nécessitent une administration de système lourde pour la bonne
coordination.
· Puissance : La grille offre de
nombreux services informatiques, qui consomment beaucoup d'énergie
électrique. Donc, alimentation sans interruption est primordiale.
2.5.8 Conclusion
Dans cette partie, nous avons vu que les besoins en puissance
de calcul pour la recherche scientifique fondamentale dépassent souvent
les possibilités qu'offre la technologie actuelle. La grille de calcul
bouleverse la façon dont les chercheurs accèdent à ces
ressources, elle reprend l'idée qu'une application lourde peut
être découpée en petites tâches isolées,
confiées à des ordinateurs différents à travers le
réseau. L'aspect économique est particulièrement
25
séduisant puisqu'il s'agit d'utiliser la puissance de
calcul et les espaces de stockage inutilisés des ordinateurs d'un
immense parc informatique. La technologie de grille de calcul a
prouvéqu'elle est la meilleure technologie pour travailler sur divers
domaines : le commerce, les entreprises, formations, la science, la recherche
et le développement. La virtualisation élimine les limitations
géographiques et économiques des ressources. Elle aide les grands
projets à accomplir en peu de temps. Cette nouvelle technologie
élimine la dépendance de projet sur un serveur principal ou super
calculateur. Pourtant, la techno-
logie de grille a besoin de se concentrer sur les questions
de sécuritéet de confidentialitéà travers les
connexions Internet.
26
2.6 Portail GVSS
Dans la découverte de médicaments, la
simulation de docking moléculaire est une méthode courante pour
prédire les potentiels interaction de petites molécules sur des
sites de liaison
de protéines. Cependant, la recherche de tous les
conformations optimales d'un composépourrait être un processus
long et onéreux. GVSS est un service pour le criblage virtuel
protéines-ligands à graned échelle
in-silico, il fournit un système de production pour
accélérer le processus de recherche de nouveaux
médicaments. Ce service de docking in-silico profite des
services de la technologie de grille de calcul, afin de raffiner la
découverte de médicaments. En outre, ces activités
facilite également plus d'applications biomédicales e-Science en
Asie.
2.6.1 Introduction
Depuis le premier défi de données mondial de la
grippe aviaire 2005, l'Academia Sinica Grille Centre de Calcul (ASGCC),
au sein de la collaboration EGEE, a étéconsacrée
àl'élaboration et le raffinage de criblage virtuel
pour les maladies négligées et émergentes
telles que la grippe aviaire, la fièvre dengue, etc.
La simulation de docking moléculaire est un processus qui prend du temps
pour une recherche exhaustive de toutes les conformations possibles d'un
composé. Toutefois, le processus massif in-silico
bénéfice du haut débit de la technologie de la grille
de calcul. Fournissant une puissance de calcul intensif et une gestion efficace
des données, l'e-infrastructure (EUAsia VO) pour la découverte
in-silico de médicaments pour les maladies
épidémique en Asie.
GAP (Grid Application Platform) et GVSS (Grid
enabled Virtual Screening Services) ont
étédéveloppés avec le moteur de docking d'AutoDock
3.0.5. GAP est un environnement de développement d'applications de haut
niveau pour la création de services d'application de la grille [7]. GVSS
est une interface graphique utilisateur de type Java, qui a
étéconçue pour la conduite de docking moléculaire
à grande échelle plus facilement sur l'environne-ment de grille
de gLite [7]. Les utilisateurs finaux utilisent GVSS sont autorisés
à spécifier la cible et la bibliothèque de
composés, mis en place des paramètres de docking, surveiller les
jobs de docking et les ressources informatiques, visualiser et affiner les
résultats de docking, et enfin de télécharger les
résultats finaux. Il existe d'autres enjeux à encourager les
activités biomédicales et intégrer plus davantage de
ressources dynamiques pour soutenir la simulation de criblage virtuel à
grande échelle en Asie. Par exemple, les scientifiques étudient
la nouvelle structure cible, par conséquent, il/elle doit savoir comment
modéliser la cible et la préparer en utilisant AutoDockTools.
On aurait aussi besoin d'une interface utilisateur conviviale pour
rejoindre et accéder à la collaboration, pour soumettre les jobs
de docking, suivre leur progrès, visualiser le docking et enfin analyser
les résultats.
27
Les utilisateurs préparent les fichiers de criblage
virtuel dans l'interface utilisateur graphique GVSS, puis sélectionnent
les ressources de la grille de calcul pour soumettre des jobs. Ces jobs
informatiques sont gérés par GAP/DIANE pour distribuer les agents
de grille de calcul à la grille [18]. Les résultats de calcul
sont gérés par AMGA , qui est un catalogue de
méta-données pour stocker des éléments de stockage
[16].

FIGURE 11 - Portail GVSS
(http: // gvss2. twgrid. org/ )
Pour faire le docking moléculaire à grande
échelle qui fonctionne sur l'environnement de la grille, ASGC a
développél'application GVSS (Grid enabled Virtual Screening
Services) qui intégre l'intergiciel gLite DIANE2/GANGA et
AMGA d'EGEE. Toutes les tâches informatiques sont gérés par
GAP/DIANE afin de distribuer les Workers de la grille de calcul. Les
résultats de calcul sont gérés par AMGA, catalogue de
métadonnées pour stocker des éléments de stockage.
GVSS utilise Autodock également en tant que moteur d'amarrage. Le GVSS a
étécréépar l'intégration de plusieurs
frameworks conçus pour des applications de grille de calcul.
2.6.2 La plate-forme GAP
GAP (Grid Application platform) est un environnement
de développement d'applications de haut niveau pour la création
des services d'application production/qualitéde grille par l'approche
MVC (Model-View-Controller) [7]. Il divise l'espace de
développement d'appli-cation de la grille en trois grandes étapes
: le portage d'application »gridification», concep-
tion de workflow de job complexe et interface utilisateur
personnalisé. Correspondant àces trois stades de
développement, le système GAP est composéde trois
sous-frameworks,
respectivement : le framework de base, le framework
d'application, et le framework de présentation.
·
28
Le framework de base fournit une couche d'abstraction à
l'interface de l'environ-nemnt distribuésous-jacente des ressources
informatiques. Il cache les complexités techniques de la gestion des
utilisateurs et des jobs de calcul en isolant les détails de mise en
oeuvre en vertu d'un ensemble d'API Java bien défini. Avec la conception
orientée objet, le framework de base a étéétendue
pour intégrer une interface de gestion de job de haut niveau
appeléDIANE.
· Le framework d'application introduit une approche
basée sur l'action pour le développement de flux de travail
»workflow» avancéet des applications complexes pour
les problèmes scientifiques réels. En utilisant les API de
framework de base, les développeurs d'applications sur cette couche
peuvent se concentrer sur la conception de workflow sans se préoccuper
des détails et/ou des modifications de l'environne-ment informatique sur
lequel les jobs informatiques seront exécutés.
· Contrairement aux framework de base et d'application,
le framework de présentation de GAP est librement défini, alors
une libertéde choix pour les applications d'adop-ter leur technologie
d'interface préférée basésur Java (par exemple,
portail Web, interface graphique, etc).
2.6.3 Architecture GVSS
Dans le service GVSS, AMGA est utilisépour
gérer l'indexation et les résultats d'amar-rage répartis.
Basésur le workflow d'analyse de données, un ensemble de
métadonné-es de la bibliothèque de composés, les
protéines cibles, et les résultats d'amarrage sont soigneusement
conçus par des biologistes participants à la mise en oeuvre. Pour
mettre en place le service GVSS, le framework DIANE a
étéintégrépour la gestion des jobs
distribués. La façon de présenter et de gérer les
jobs sur la grille est entièrement contrôlée par ce
framework. Le développement de cette interface permet de réduire
l'effort pour communiquer avec l'environnement de la grille. Une application
graphique en Java a étédéveloppépour les
utilisateurs finaux afin d'utiliser les services de GVSS. Le
développement de cette interface profite des avantages des frameworks de
base et d'application de GAP pour réduire l'effort de communiquer avec
l'environnement de grille de calcul.

29
FIGURE 12 - Architecture Service de criblage
virtuel GAP (GVSS) [7]
2.6.4 Conclusion
GVSS est développépour prédire comment
les petites molécules interagissent avec le récepteur. Il
réduit considérablement le coût en utilisant la demande
dynamique des ressources de la grille de calcul. Le portail GVSS facilite la
découverte de médicaments en permettant aux utilisateurs un
accès simultanéet instantanéaux ressources de la grille,
tout en masquant la complexitéde l'environnement de la grille aux
utilisateurs finaux.
30
2.7 Plate-formes utilisés
2.7.1 WISDOM
WISDOM (Wide In Silico Docking On Malaria) est une initiative
qui a étélancéen 2005 pour utiliser les nouvelles
technologies de l'information et dépolyer des applications de docking de
grande échelle, afin de chercher et de découvrir des
médicaments contre le plaudisme et d'autres maladies dites
négligées. Le but de WISDOM est de prouver la pertinence
de l'utilisation de la grille de calcul dans la recherche de médicaments
et de traitement pour les maladies dangereuses [8]. Il travaille en
étroite collaboration avec EGEE, et il fait usage de l'infrastructure
EGEE pour exécuter un grand nombre de données. WISDOM est
considérécomme une première étape pour mettre en
place une recherche de médicaments in-silico sur une
infrastructure de grille. La plate-forme WPE (WISDOM Production Environment),
développépar LPC (Laboratoire Clermont Ferrand-France), a
étéutiliséavec succès pour le projet WISDOM dans la
découverte de nouveaux inhibiteurs contre le Malaria. Cette plate-forme
fournit une couche entre les utilisateurs et l'environ-nement de la grille de
calcul afin de dissimuler sa complexité. Avec cette plate-forme les
utilisateurs peuvent facilement utiliser les ressources de la grille pour
effectuer leur calcul.
2.7.1.a Définition WISDOM
WISDOM est un intergiciel conçue comme un
environnement de gestion de l'expérience. Il gère les
données, les jobs, et partage la charge de travail sur toutes les
ressources intégrées, même si elles adaptent
différentes normes technologiques. Il est tout a fait possible de
construire des services web qui interagissent avec le
système. WISDOM est considérécomme un ensemble de services
génériques agissant comme un niveau d'abstraction pour
les ressources et offrant une gestion générique
des données et des jobs de sorte que les services d'applications peuvent
utiliser l'un des services sous-jacents d'une manière très
transparente [11]. L'initiative WISDOM comprend trois objectifs, l'objectif
biologique, qui consiste à proposer de nouveaux inhibiteurs pour une
famille de protéine produite par plasmodium, l'objectif
biomédical, qui repose sur le déployement d'une application de
docking in-silico sur une infrastructure de grille de calcul, et
l'objectif de grille, qui s'ap-puie sur le déploiement d'une application
très demandeuse en temps de calcul et générant une grande
quantitéde données pour tester l'infrastructure de grille et ses
services. Les utilisateurs ne sont pas en interaction directe avec les
ressources de la grille, et ils ne sont pas censés de savoir comment
cela fonctionne, car ils sont juste en interaction avec les services de haut
niveau, tout comme avec un autre service web.
31
2.7.1.b Architecture WPE
L'environnement de production WISDOM (WPE) est
considérécomme un intergiciel installésur des ressources
de calcul pour gérer des données et des jobs et pour partager la
charge sur l'ensemble des ressources intégrées [24]. Il est
possible de construire des services web qui interagissent avec le
système. Les quatre composants principaux de WPE (WISDOM Production
Environment) sont [9] :
· Le gestionnaire des tâches »Task
Manager» interagit avec le client et accueille les tâches
créées par le client.
· Le gestionnaire de jobs »Job Manager»
soumet des jobs aux éléments de calcul (CE), du sorte que
les tâches gérées par le gestionnaire des tâches
seront executées.
· Le système d'information WIS »WISDOM
Information System» utilise AMGA »ARDA Metadata Grid
Application», pour stocker toutes les métadonnées
requises pour le gestionnaire de job.
· Le gestionnaire de données »Data
Manager», gère les fichiers sur la grille de calcul.

FIGURE 13 - Architecture WPE [9]
Tout d'abord, le module de gestionnaire de job »Job
Manager» reçoit les demandes et soumet les jobs pilotes
»pilot agents» sur la grille de calcul, afin de
réaliser des tâches dans le gestionnaire des tâches
»Task Manager». L'exécution de gestionnaire de job
nécessite un certificat qui correspond à l'organisation virtuelle
oùles jobs seront soumis. Ensuite, les tàaches sont
enregistrées et gérées par les gestionnaire des
tàaches. Un agent interagit avec le gestionnaire de tàache pour
récupérer une tàache et l'exécute sur la grille de
calcul. Après, le module WIS »WISDOM Information System»
enregistre les états des agents et contrôle l'information des
agents de pilotes sur la grille. Et le gestionnaire de données
gère les fichiers sur la grille en mode batch.
32
2.7.1.c Conclusion
Le but de WISDOM est de prouver la pertinence de
l'utilisation de la grille de calcul dans la recherche de médicaments
pour les maladies négligées, et de produire une grande
quantitéde données dans un temps
limitéavec un faible coût, tout en avoir recours
àl'infrastructure et les ressources de la grille de calcul.
En dépit de son utilisation réussie pour les données de
grand échelle, la plate-forme est confrontée aux certaines
limites :
· Son service d'information est ralenti après un
long temps d'exécution.
· Les agents de pilotes sont tués pour des raisons
inconnues.
· La plate-forme WPE s'arrête lorsque le
gestionnaire des tâches est vide pendant une longue période.
33
2.7.2 DIRAC
DIRAC (Distributed Infrastructure with Remote Agent Control)
est un framework logiciel pour le calcul distribué[19]. Il a
étédéveloppépour l'expérience en Physique
des Hautes Energies LHCb au LHC, CERN. DIRAC fournit une solution
complète pour une ou plusieurs communautés d'utilisateurs, qui
demandent un accès aux ressources distribuées. Il construit une
couche entre les utilisateurs et les ressources de la grille de calcul, tout en
offrant une interface commune à un nombre de fournisseurs
hétérogènes, en les intégrant
de façon homogène, fournissant une
interopérabilité, une transparence et une
fiabilitéd'utilisation de ressources.

FIGURE 14 - Intergiciel
DIRAC
(http: // diracgrid. org/
)
2.7.2.a Architecture et composants DIRAC
L'architecture modulaire du DIRAC est conçue pour une
extension aisée en fonction des besoins des applications
spécifiques, et qui suit le paradigme de l'architecture orientée
services (SOA). Les composants de DIRAC peuvent être groupés en 4
catégories : ressources, services, agents et interfaces.
1. Service
Tous les services de DIRAC sont écrits en Python et
implémentés en tant que serveurs XML-RPC. La bibliothèque
standard de Python fournit une implémentation complète du
protocole XML RPC pour le serveur et une partie du client.
2. Élément de calcul »Computing
Element»
L'élément de calcul (CE) dans DIRAC est une API
faisant abstraction à des opérations courantes de manipulation de
job par les systèmes informatiques de traitement par lots (batch). Il
permet également d'accéder à des informations
d'état de ressource de calcul.
3. 34
Système de gestion de la charge de
travail
Le système de gestion de la charge de travail
(Workload Management System) se compose de trois
éléments principaux : service central de gestion de job (JMS),
agents distribués en cours d'exécution près des
éléments de calcul de DIRAC et gestionnaire de job. JMS est
à son tour un ensemble de services qui fournit la réception et le
tri des jobs dans les files d'attente de tâches, servant des jobs aux
demandes de l'agent, l'accumulation et le service d'information de
l'état du job. Les agents vérifient en permanence la
disponibilitédes éléments de calcul (CE), retire les jobs
du JMS et oriente l'exécution du job au ressource informatique locale.
Le gestionnaire des jobs (Job Wrapper) préparent
l'exécution du job sur le noeud de travail (Worker Node),
récupère l'entrée (le sandbox) du job, envoie des
informations d'état des jobs au JMS, et
télécharge la sortie du job. Les jobs sont décrits en
utilisant le Job Description Language (JDL).
4. Système de gestion des
données
Le système de gestion des données »Data
Management System», inclut les services de fichier de catalogue, qui
gardent la trace des ensembles de données disponibles et leurs
répliques, ainsi des outils d'accès aux données et la
réplication.
· Fichier catalogue
Une base de donnée qui assure le suivi des jobs
exécutés et les métadonnées des jeux de
données disponibles, maintient également des informations sur les
répliques physique des fichiers.
· Élément de stockage
L'élément de stockage (SE), est une combinaison
d'un serveur standard, comme GridFTP, et les informations stockées dans
le service de configuration sur la façon d'y accéder. L'API de SE
offre la possibilitéde brancher dynamiquement des modules de protocole
de transport par lequel le SE est accessible.
· Service de transfert de fichier
Le transfert de fichiers est une opération fragile en
raison des échecs ou des erreurs matérielles de réseau et
de stockage potentiels dans les services logiciels associés. Par
conséquent, un service de transfert de fichier fiable (RFTS) permet de
nouvelle tentative des opérations ayant échouéjusqu'au
succès total.
35
5. Service de configuration
Le service de configuration (CS) fournit des
paramètres de configuration nécessaires
à d'autres services, afin d'assurer la collaboration
entre les agents et les jobs.

FIGURE 15 - Architecture DIRAC [20]
La plate-forme DIRAC permet le déploiement des agents
de pilotes sur les Worker Node comme des jobs réguliers
à l'aide de mécanisme de planification de grille [21]. Tout
d'abord, l'utilisateur crée une tâche. Ensuite l'agent pilote est
soumis sur la grille pour exécuter cette tâche.
La soumission des jobs sur la grille de calcul avec
l'intergiciel DIRAC, nécessite un langage spéciale
»JDL». JDL signifie Job Description Language, il
est le moyen standard de description de job dans l'environnement de grille
de calcul. Ci-dessous, un exemple d'un script jdl qui permet de soumettre un
job de docking sur la grille de calcul.
JobName = "testJob-biomed";
Executable = "
dock1.sh";
StdOutput = "std.out";
StdError = "std.err";
InputSandbox = {"
dock1.sh",
"LFN:/biomed/user/l/louacheni/
file.tar.gz"};
OutputSandbox = {"std.out","std.err", "fileDock1.tar.bz2",
"fileDock2Aug.tar.bz2"};
OutputSE = "DIRAC-USER";
36
2.7.2.b Conclusion
Le projet DIRAC est un produit universel largement
utilisépar différents partenaires, dans différents
contexte, et qui permet de construire des systèmes de calcul
distribués en utilisant les différentes ressources de calcul
comme ordinateurs individuel, les clusters ou bien les grilles de calcul. La
structure modulaire de DIRAC permet de l'adapter rapidement aux besoins
particuliers des différentes communautés d'utilisateurs pour
faciliter leur accès aux ressources et services de la grille de calcul.
Les principaux avantages de DI-RAC sont : simplicitéd'installation, de
configuration, fonctionnement des divers services et sa capcacitéde
gérer une grande quantitéde ressources de données. Et son
objectif est à savoir aider les utilisateurs à communiquer
facilement avec l'environnement de la grille, soumettre, contrôler et
surveiller leur job. Pour ces raisons nous avons choisit DIRAC pour en faire
partie dans la réalisation du portail.
Les plate-formes WPE et DIRAC ont le
même objectif, à savoir aider les utilisateurs
àcommuniquer facilement avec l'environnement de la grille de
calcul, soumettre, contrôler
et surveiller les jobs. Cependant, leurs architectures sont
très distinctes. En premier lieu, pour la plate-forme WPE un
agent de pilote est soumis sur la grille. Ensuite, l'utilisateur crée
une tâche dans le gestionnaire des tâches. Et un agent de pilote
WPE exécute de nombreuses tâches. Tandis que pour la
plate-forme DIRAC, l'utilisateur crée une tâche. Ensuite,
un agent de pilote est soumis à la grille pour l'exécution de
cette tâche [8]. Et un agent de pilote DIRAC exécute une
tâche.
37
3 Implémentation
Le but de ce travail est de déveopper un portail web
pour le criblage virtuel, qui permet aux utilisateurs (biologistes, chimistes,
bio-informaticiens...) d'envoyer des jobs de docking sur la grille de calcul et
de récupérer les résultats à travers ce portail,
dont le but accélérer
la recherche de médicaments. Afin de faciliter la
tâche aux utilisateurs nous avons proposéune architecture pour le
portail que nous allons la détailler ci-après. Oùplusieurs
outils et technologies ont étédéployés afin de
mener à bien ce projet.
3.1 Architecture du système proposée
Nous avons proposéune architecture simple afin
d'offrir aux utilisateurs finaux qui ne sont pas des experts ni en
informatique, ni en technologie de la grille, une interface conviviale est
facile à utiliser sans qu'ils se préoccupent de la
complexitédu portail. Outre les solutions basées sur le Web.
Notre solution repose sur l'utilisation de Taverna [6], qui est un outil
développépar le consortium myGrid, permettant la
réalisation de traitements in-silico sous la forme de
workflows, tout particulièrement dans le domaine de bio-informatique.
Cet outil permet l'exécution des expériences scientifiques dans
la forme de workflow. Chaque workflow est constituépar une série
de services reliés l'un à l'autre. Taverna est conçu pour
combiner des services web distribués et/ou des outils locaux dans des
pipelines d'analyse complexes, afin de réaliser la conception et
l'exécution des workflows scientifiques. Nous avons
utilisél'intergiciel DIRAC, qui va servir comme intermédiaire
entre le portail et les ressources de la grille pour la soumission des jobs de
docking sur la grille de calcul, et la récupération des
résultats à partir de l'espace de stockage de la grille. Et nous
avons eu recours à l'outil AutoDock, pour effectuer le docking
moléculaire protéine-ligand. Cependant, cela nécessite
l'installation de ces outils afin de pouvoir profiter des avantages de Taverna,
DIRAC et AutoDock.
Le schéma ci-dessous illustre l'architecture que nous
avons utilisépour implémenter le portail web pour le criblage
virtuel. L'utilisateur accéde au portail web en s'authentifiant avec ses
credentials valides, il envoie sa requête pour effectuer l'amarrage,
après les paramètres d'entrées au Client Taverna. Ce
dernier récupère le worflow Taverna, qui contient les services
web nécessaire pour le docking (génération et
soumission des jobs sur la grille, suivre l'état du job soumis et
récupération de résultat). Après avoir
récupérer le workflow, le client Taverna exécute les
services pour le docking. L'un des services web, est de générer
les fichiers »JDL», et à soumettre les job sur la
grille de calcul en utilisant l'intergiciel DIRAC. Le serveur DIRAC
soumet les jobs de docking générés par Taverna service sur
l'élément de calcul de la grille. Les données
stockées sur les éléments de stockage sont alors
transférés sur le noeud de calcul, puis les résultats sont
stockés sur un élément de stockage de la grille, et
répliqués sur d'autres éléments pour
réaliser une copie de sauvegarde. À la fin, l'utilisateur peut
télécharger les résultats du docking

38
FIGURE 16 - Architecture du système
proposée
3.2 Outils utilisés
Comme illustre l'architecture du système ci-dessus,
nous avons utiliséplusieurs outils : AutoDock, pour préparer les
fichiers nécessaires afin d'effectuer le docking moléculaire
protéine-ligand. Pour atteindre l'objectif de ce travail, nous avons
utiliséla Grille EGI,
via la VO Biomed et l'intergiciel DIRAC, oùun ensemble
de scripts avaient déjàétédéveloppé,
nous avons choisi d'adapter ces scripts sur la grille via DIRAC. L'adaptation
a
principalement consistéà générer
les fichiers JDL et à utiliser les commandes de DIRAC :
»dirac-wins-job-subinit» pour soumettre les jobs sur la
grille de calcul. Les identifiants des jobs soumis ont
étéstockés dans un fichier local, qui a ensuite servi
à tester le statut des jobs avec la commande
:»dirac-wins-job-status», et à
récupérer les résultats avec :
»dirac-wins-job-get-output». La visualisation du workflow
s'est fait avec »Taverna», cet outil est largement
utiliédans le domaine de la bio-informatique [23], et qui permet aux
utilisateurs d' effectuer des expériences scientifiques, de visualiser
et de créer leurs workflow.
Afin de comprendre le fonctionnement de ces outils, nous
avons installéle client DIRAC (voir annexe) pour mieux cerner
le mécanisme de soumission des jobs sur la grille de calcul à
travers l'intergiciel DIRAC. Et pour l'outil Taverna [23], nous avons
installéTaverna Workbench version 2.5 (http: //
www. taverna.
org. uk/ download/ workbench/ 2-5/ )
pour le mode graphique, il permet aux utilisateurs d'identifier
et combiner des services .Et Taverna Command Line (2.5) pour le mode ligne de
commande (http: //
www. taverna.
org. uk/ download/ command-line-tool/ 2-5/
), sous le système d'exploitation Linux.
39
Pour le mode graphique, il nécessitera l'installation
d'un paquet nomméGraphviz (http: // www. graphviz. org/
) pour la visualisation du workflow. L'exécution de Taverna est
simple, il suffit juste de lancer le fichier »
taverna.sh». Afin
de montrer la création des workflow avec Taverna, nous avons choisi un
exemple qui concerne la génération de script JDL (Job
Description Language) et la soumission des jobs sur la grille de calcul
avec DIRAC via la commande (dirac-wms-job-submit). Ce workflow accepte
comme entrées deux fichiers essentiels (fichiers »dpf»
& »gpf»), et la sortie est l'identifiant du job soumis sur
la grille. son statut, et le résultat du job.
Nous avons exécutéle workflow avec le mode
graphique comme c'est apparaît dans la capture ci-dessous :

FIGURE 17 - Workflow soumission de job sur la
grille avec Taverna
Le résultat de ce workflow est l'identifiant du job
comme montre la capture ci-dessus (JobID = 18473187), un dossier qui
contient les fichiers générés par le workflow (script
»jdl» et »shell»), l'état du job, et le chemin
du résultat du job après l'avoir récupérer depuis
la grille de calcul. Et en mode ligne de commande en utilisant Taverna
Command Line avec la ligne de commande suivante:
40
> /bin/bash taverna-commandline-core-2.5.0/
executeworkflow.sh -inputvalue
file tvrnCmdJdl
-inputvalue gpf 1OKE.gpf
-inputvalue dpf ZINC71389186 01 1OKE.dpf
/home/dida/Documents/TavernaStage/jobStatOut.t2flow

Nous avons choisi l'outil Taverna dans notre architecture, vu
son utilisation pour concevoir des workflows scientifques et exécuter
des expérimentations »in-silico»,
possibilitéd'importer des services via URL, d'un
côté, et qu'on pourra dans le future étendre le workflow
tout en ajoutant d'autres services et d'autres fonctionnalités afin de
répondre
aux exigences des utilisateurs, de l'autre côté.
Et en ce qui concerne l'intergiciel DIRAC,
nous l'avons choisi car il procure les avantages suivants :
· Accessibilitéet simplicitéde l'interface
d'utilisateur
· Lignes de commandes très enrichit
· Efficacitéd'exécution des tâches
élevée
· Exécution rapide des tâches des
utilisateurs
· Soumission des jobs en parallèle
· Production des données massives
· Possibilitéd'inclure des ressources de calcul
supplémentaires
· Intégration des ressources informatiques
hétérogènes
· Stabilitéd'agent de pilote
· Réduction de temps de réponse
· Récupération rapide du résultat du
job
41
3.3 Conception du portail
Cette partie est consacréà l'étude
conceptuelle de notre projet, oùnous présentons les
différentes étapes réalisées durant
l'implémentation de notre portail. Nous commençons par
décrire la conception de la base de donnée, ensuite nous
définissons les fonctionnalités et les services de notre
portail.
Nous avons élaborénotre base de donnée
pour le portail avec MySQL. Cinq tables font partie de notre conception :
· Ligand, elle contient toutes les
informations nécessaire concernant le ligand. L'at-tribut file name
est le fichier que l'utilisateur l'a
déjàpréparéavec AutoDockTools, et qui sera utiliser
lors du docking.
· Protein, cette table contient les
informations à propos du protéine. Elle un attribut important qui
sera utiliser dans le docking. Il s'agit de file name, qui est un
fichier de protéine que l'utilisateur l'a
déjàpréparéavec »ADT».
· Map Parameter, oùl'attribut
file tar gz contient les fichiers de paramètres de la grille,
que l'utilisateur les a déjàpréparéavec
autogrid4.
· Project, cette table contient tout les fichiers
nécessaires qui seront utilisés pour lancer des jobs de docking
sur la grille de calcul. L'attribut file dpf est le fichier de
paramètres de docking préparer par l'utilisateur avec
AutoDockTools.
· User, cette table contient les
credentials des utilisateurs enregistrés.
Afin de faciliter la tâche aux utilisateurs, nous avons
ajoutéune table pour les paramètres de grille. Au lieu de faire
entrer manuellement les coordonnées (X,Y et Z) des paramètres de
la grille, l'utilisateur ne doàýt que préparer les
fichiers des paramètres avec l'outil AutoDockTools.
La conception de la base de donnée pour notre portail
web est présentédans le schéma ci-dessous.
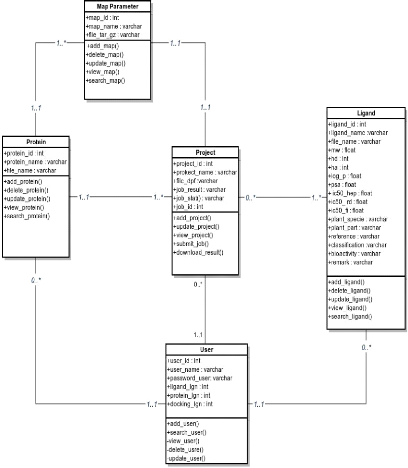
42
FIGURE 18 - Diagramme de classe du portail
web
mw : poids moléculaire hd : donneur
d'hydrogène ha : accepteur d'hydrogène log p : coefficient de
partition
· Cas d'utilisation
Les diagrammes ci-dessous présentent les cas
d'utilisation, afin de montrer les différentes interactions entre les
utilisateurs et le système. 4 types d'utilisateurs sont présents
dans notre conception, celui du ligand, du protéine, du docking et
l'administrateur du site.
Le premier cas d'utilisation appartient au Ligand.
Oùl'utilisateur peut ajouter, modifier, supprimer un Ligand, mais cela
nécessite une authentification de l'utilisateur s'il possède un
compte, sinon il do^~t s'enregistrer avant d'effectuer ces opérations.
Tous les utilisateur peuvent consulter le catalogue des ligands disponibles,
voir les informations concernant ces ligands et chercher un ligand.
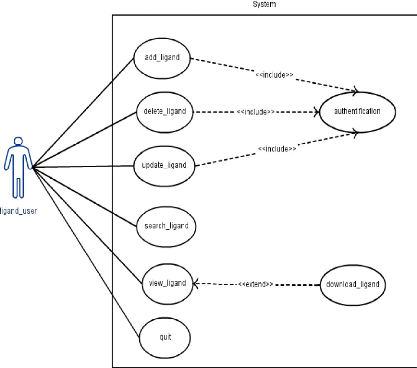
43
FIGURE 19 - Cas d'utilisation pour le
Ligand
Le deuxième cas d'utilisation concerne la
molécule de protéine. Les utilisateurs authentifiés sont
capables d'effectuer les opérations d'ajout, suppression et
modification. Et les autres utilisateurs non-authentifiés peuvent
consulter la liste des protéines disponibles sur le portail, et
d'effectuer une recherche sur une protéine.
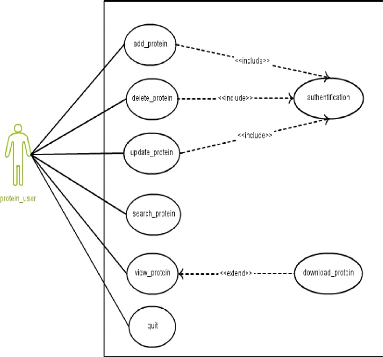
44
FIGURE 20 - Cas d'utilisation pour la
Protéine
Le troisième cas d'utilisation appartient aux
paramètres de grille (map parameter). Tel que, une
protéine peut avoir plusieurs paramètres de grille. Les autres
utilisateurs qui ne possèdent pas de compte peuvent consulter la liste
des paramètres de grille.
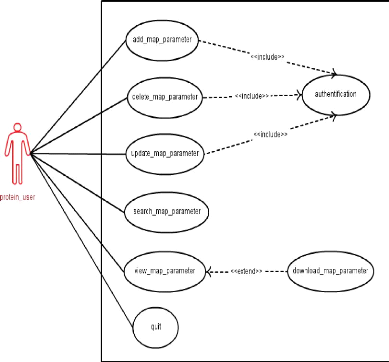
45
FIGURE 21 - Cas d'utilisation pour les
paramètres de grille
L'avant dernier cas d'utilisation, est le cas crucial dans
notre projet. Il s'agit d'effec-tuer l'amarrage. L'authentification des
utilisateurs est requises pour pouvoir créer un projet, soumettre le job
de docking et télécharger les résultats. Tout d'abord
l'utilisateur accède au portail, il s'authentifie s'il a l'intention
d'effectuer le docking. Puis il crée un projet, en choisissant les
fichiers dont il a besoin pour réaliser l'amar-rage, et après il
soumet son job sur la grille de calcul. À la fin, il peut
récupérer le résultat du job en
téléchargeant le fichier de docking.
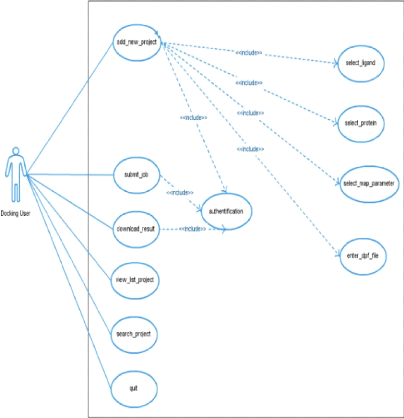
46
FIGURE 22 - Cas d'utilisation pour le
docking
47
Le dernier cas d'utilisation concerne l'administrateur du
site. Ce dernier possède les droits de gérer la liste des
utilisateurs, des ligands, des protéines, des paramètres et les
projets de docking.
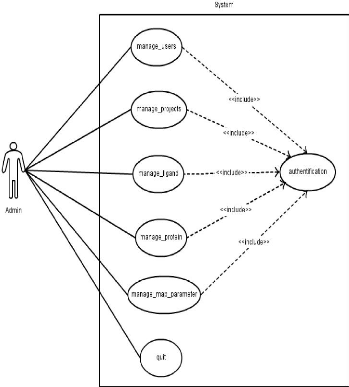
FIGURE 23 - Cas d'utilisation pour
l'administrateur du portail
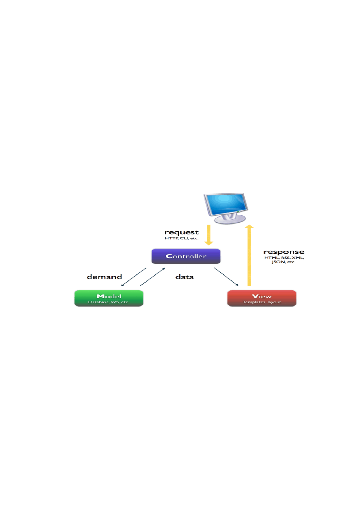
3.4 Développement du portail du web
Nous avons eu recours à plusieurs technologies du web
pour le développement du portail. Parmi ces technologies : PHP comme
cadre de base et les services web JAX-WS avec JAVA pour interagir avec le
système du portail, tout en recevant les requêtes du client. Le
portail interagit avec le système via l'intermédiaire du web
service. Notre application est réalisée sous Linux et avec
Netbeans 8.0. Nous détaillons, dans ce qui suit, chacun des outils et
langages utilisés pour la manipulation des données ainsi pour
l'implémentation du portail web.
Le portail web a étédéveloppéen
utilisant le framework Yii (Yes It Is), qui est un framework PHP
basésur des composants ultra performant qui a
étédéveloppépour créer des application Web
de grande qualité, dont le but d'accélerer le
développement des applications Web. Yii est développéen
respectant le modèle MVC (Model-View-Controller).
FIGURE 24 - Modèle MVC
3.4.1 Les services web
Nous avons développétrois services web en Java
sous NetBeans 8.0 avec le serveur GlassFish
et Apache Axis2. Chaque service est responsable à une
fonctionnalitépour notre projet. Le premier service prend en
entrée les fichiers dpf & gpf , pour
générer les fichiers nécessaires (jdl et
shell), et il soumet les job de docking sur la grille de calcul via DIRAC
à partir des fichiers jdl qu'il les a générer. Le
deuxième service prend en entrée l'identifiant du job
(jobID) qu'il a soumet, et donne comme sortie l'état du
job. Si l'état du job est à (Failed), alors le
service re-programme le job et il re-soumet à nouveau le job avec la
commande de DIRAC (dirac-wins-file-reschedule jobID). Le dernier
service repose sur la récupération du résultat du job
depuis la grille de calcul. Il prend en entrée l'identifiant
»jobID» et l'état du job.
48
49
Si le statut du job est à l'état (Done),
alors le service récupère le résultat du job
àpartir de l'espace de stockage de la grille de calcul.
Après avoir exécuter le service web sous Netbeans, un script WSDL
(Web Service Description Language) a
étégénéré. L'URL de WSDL sera
utilisépour créer des workflows avec l'outil Taverna.
Le tableau ci-dessous présente en détail les
services web que nous avons implémenté, (les paramètres
d'entrées, les sorties et une description du rôle de chacun).
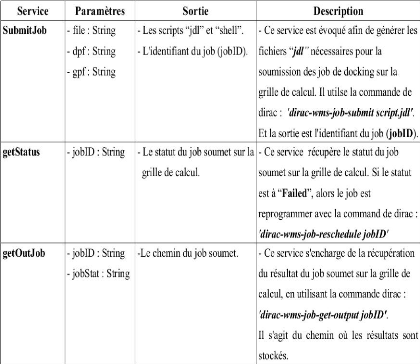
FIGURE 25 - Description des services web
implémentés
- Le service »submitJob», ce
service récupère les fichiers d'entrées à partir du
portail, puis il génère les scripts »jdl» dans
un dossier. Après la génération des scripts, les jobs sont
soumis sur la grille de calcul à travers l'intergiciel
»DIRAC» en utilisant la commande
»dirac-wins-job-subinit».
- Le service »getStatus», le
rôle de ce service est de suivre l'état des jobs soumis sur la
grille avec la commande »dirac-wins-job-status». Si
l'état du job est à»Failed»,
alors le service re-programme la soumission du job avec la commande de
DIRAC »dirac-wins-job-reschedule».
50
- Le service »getOutJob», sert
à récupérer le résultat du docking à partir
de la grille via la commande DIRAC »dirac-wins-job-get-output»
si et seulement si l'état du job est à
»Done».
Toutes les opérations de service (SubmitJob,
getStatus, getOutJob) ont ététestées en utilisant Taverna.
Et chaque opération dispose de son propre workflow. Nous passons
à l'étape crucial de notre travail, et qui repose sur le
déployant de l'outil Taverna, qui est un outil très
utilisépour créer et visualiser des workflows scientifique. Le
workflow a étémis en oeuvre en utilisant l'adresse de service
(http: // localhost: 8080/ TavernaWS/ WSSubmitJob? Tester) avec l'outil
Taverna Workbench, pour être exécutéà
travers l'intergiciel »DIRAC».
51
La figure suivante illustre le workflow des services web
implémentés pour le portail web avec Taverna Workbench
:
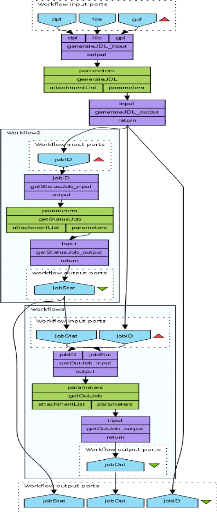
FIGURE 26 - Workflow des services web du
portail
52
4 Expérimentation & Résultats
Cette partie se focalisera sur la démonstration du
portail web et les résultats du docking. La figure ci-après
présente la fenêtre principale de notre portail. Ce portail a
étéconçu pour les utilisateurs qui ne sont pas
forcément des experts en informatique (biologistes, chimistes,
bio-informaticiens,...), pour qu'ils puissent effectuer le criblage virtuel
in-silico sur la grille de calcul. Les visiteurs peuvent
accéder au portail afin de consulter la liste des ligands,
protéines, les paramètres de grille et les projets qui ont
étédéjàsoumet sur la grille. Mais s'ils veulent
effectuer une tâche ils doivent, tout d'abord, s'enregistrer ou
s'authentifier en fournissant leur nom et leur mot de passe.
FIGURE 27 - Interface d'accueil du portail
web
L'utilisateur accède au portail par une simple
authentification (nom & mot de passe). La figure ci-dessous montre
l'interface pour s'enregistrer auprès du portail afin de profiter de ses
services. L'utilisateur entre son nom et son mot de passe, et il doit choisir
sa catégorie (soit ligand, soit protéine ou docking).
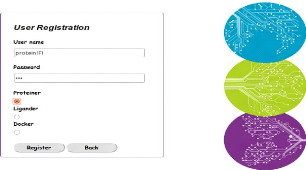
FIGURE 28 - Interface de création d'un
nouveau compte
Après la phase d'enregistrement, l'utilisateur do^~t
s'authentifier pour bien profiter des privilèges qu'un visiteur normal
ne possède pas.

53
FIGURE 29 - Interface d'authentification
L'administrateur du site possède tous les droits pour
la gestion du site. Il gére les utilisateurs, les ligands, les
protéines, les paramètres de la grille et les projets de
docking.
La figure ci-dessous sollicite la liste des utilisateurs.
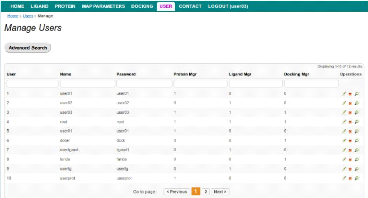
FIGURE 30 - Interface de gestion des utilisateurs
Après avoir créer un compte, l'utilisateur peut
accéder au portail en fournissant ses credentials pour accomplir sa
tâche. Si ce dernier possède un compte, et il appartient à
la catégorie de »ligand», alors il peut ajouter,
modifier son ligand, et il peut le supprimer. La capture d'écran
ci-dessous montre l'ajout d'une nouvelle molécule de ligand par
l'utilisateur.
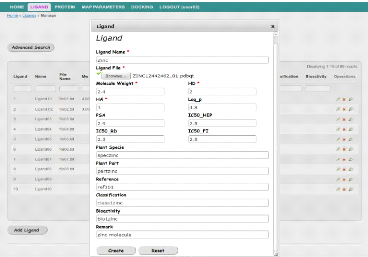
FIGURE 31 - Interface d'ajout d'un nouveau Ligand
54
Les autres visiteurs du portail non-authentifiés
peuvent consulter la liste des ligands disponibles sur le portail et effectuer
une recherche sur une molécule de ligand.
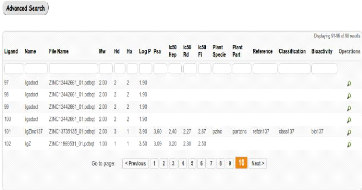
FIGURE 32 - Interface de liste des Ligands disponibles
L'administrateur de protéine peut gérer la
liste des molécules de protéines, tout en effectuant les
opérations d'ajout, de modification et de suppression. Ci-dessous, une
figure qui montre la fenêtre pour la gestion des protéines.
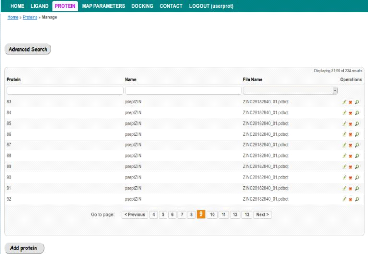
FIGURE 33 - Interface de gestion des protéines
55
56
La modification d'une protéine nécessite
l'authentification de l'administrateur de protéine, comme c'est
illustrédans la capture ci-après.
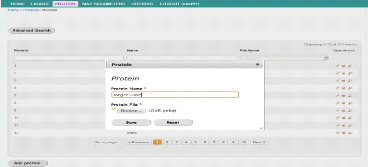
FIGURE 34 - Interface de modification d'une
protéine
Comme nous avons citédans la partie
implémentation, la relation entre la protéine et les
paramètres de la grille est une relation (1 :n). Tel que, une
protéine peut avoir plusieurs paramètres, mais un
paramètre de la grille appartient à une et une seule
molécule de protéine.
La capture suivante illustre l'ajout d'un fichier de
paramètre de la grille.
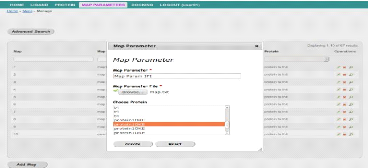
FIGURE 35 - Interface d'ajout de fichier de
paramètres de la grille
On arrive à la partie importante dans ce projet, elle
consiste à créer un projet pour le docking et soumettre les jobs
de docking sur la grille de calcul. Afin de pouvoir créer un nouveau
projet, l'utilisateur doit s'authentifier en fournissant ses credentials (son
nom et son mot de passe), sinon il do^~t créer un compte. Après
avoir authentifier, l'utilisateur est capable de créer un nouveau
projet, pour cela il do^~t choisir un ligand, une protéine, un fichier
de paramètre et aussi le fichier de paramètres pour le docking
(dpf). Pour ces paramètres d'entrées sélectionnés,
soit il les a déjàpréparer lui même, ou par d'autres
utilisateurs. La capture ci-après illustre la création d'un
nouveau projet pour le docking.
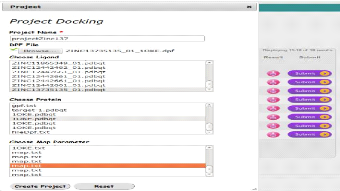
57
FIGURE 36 - Interface d'ajout d'un nouveau projet de docking
En retournant à la liste des projets qui ont
étécréés, on peut vérifier que le projet a
étéajoutéavec succès.
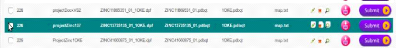
FIGURE 37 - Vérification d'ajout du nouveau projet
Après avoir créer un projet et choisir les
fichiers nécessaires pour effectuer le docking. L'utilisateur n'a
qu'appuyer sur le bouton »submit» pour soumettre son job sur
la grille. Le bouton va récupérer les fichiers à partir du
portail, puis, il fait appel à Ta-verna client qui
récupère le workflow contenant les services web. Après, il
soumet le job sur la grille de calcul. Nous allons présenter les
résultats que nous avons obtenus lors de soumission des jobs de docking
sur la grille, et la récupération des résultats à
partir de la grille à travers de ce portail web. Nous nous sommes servis
de la base de donnée ZINC ( http: //
zinc. docking.
org) , qui est une base de
donnée de composés disponibles pour le criblage virtuel (1OKE
pour la protéine, et ZINC pour le ligand), oùle fichier de
ligand comprend 10256 composants. Les fichiers de paramètres pour la
grille grid parameter et pour le docking dock parameter(dpf
& gpf) ont déjàétépréparé.
L'étape de docking moléculaire est réalisée
grâce au sous-programme AutoDock, qui recherche toute les solutions
d'amarrage en fonction des paramètres du fichier »dpf»
que l'utilisateur à déjàpréparer. Après
l'achèvement du docking, les résultats ont
étégénérés dans un fihier log avec
l'extension (glg & dlg). Le fichier »glg» contient les
affinités calculer entre les différentes types d'atomes de la
protéine et le ligand. Et le fichier »dlg», qui fournit les
coordonnées atomiques des 10 meilleurs positions du ligand dans le site
de la protéine, leur énergie d'in-teraction ainsi que les
différentes valeurs de l'écart quadratique moyen (Root Mean
Square Deviation ou le »RMSD»).
La figure ci-après montre la soumission de job de
docking, tout en sélectionnant le projet »projectZinc137».
Dans ce projet nous avons choisi: le fichier de paramètre de
docking ZINC13735135 01 1OKE.dpf , le ligand ZINC13735135
01.pdbqt, la protéine 1OKE.pdbqt et le fichier de
paramètre de la grille map.txt.
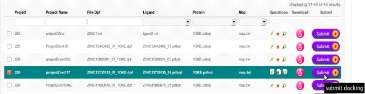
FIGURE 38 - Soumission de job de docking
Après avoir soumettre le job avec le bouton
submit, le résultat consiste en fichier zip,
oùnous l'avons spécifier dans le script jdl. Le fichier
est stokés sur l'espace de stockage de la grille de calcul, puis nous le
récupérons depuis le SE de la grille via la commande DIRAC
»dirac-dms-get-file». Ce dernier contient le fichier de
docking »dlg». La capture ci-après montre le
résultat du job stocker sur notre espace de stockage de la grille, comme
c'est illustrédans la capture ci-dessous.

Le résultat est compresséet stockésur
l'élément de stockage de la grille de calcul, puis, le
résultat est récupéréà partir de la grille
de calcul à l'aide de la commande DIRAC, pour que
l'utilisateur puisse le télécharger. La figure ci-après
montre le téléchargement du résultat du job de docking
soumis. Le fichier contient les fichiers log de docking et de grille
(»dlg» & »glg»).
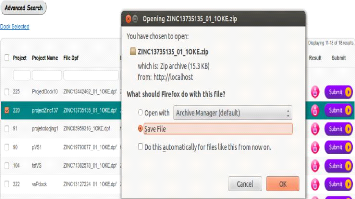
FIGURE 39 - Téléchargement du résultat de
docking
58
59
Après avoir télécharger le fichier
»zip», nous avons vérifiési l'opération
du docking a étéeffctuéavec succès, en ouvrant le
fichier log de docking. La capture ci-dessous
illustre le résultat du docking. Comme montre la
figure suivante le docking a étéachevéavec
succès.


FIGURE 40 - Fichier log de docking »dlg»
Nous avons aussi testéce portail pour soumettre un
autre job de docking du projet : ProjectZinc1OKE, qui comprend
les fichiers de paramètres suivants : fichier de paramètres de
docking (dpf) »ZINC4166-0875 01 1OKE.dpf»,
fichier ligand »ZINC41660875 01 1OKE.pdbqt», fichier
protéine »1OKE.pdbqt».

FIGURE 41 - Soumission du projet de docking
ProjectZinc1OKE
60
L'utilisateur peut récupérer son réultat
du job dès que l'opération du docking s'achèvera. Le
résultat est illustréci-après.
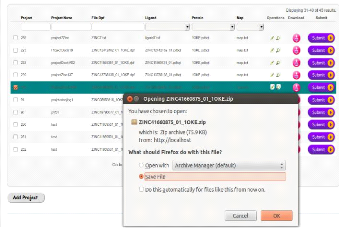
FIGURE 42 - Téléchargement du résultat de
docking
On peut voir que le résultat du docking est bien
enregistrédans l'espace de stockage de la grille de calcul,
oùnous avons compresséles fichiers résultant du
docking.

FIGURE 43 - Enregistrement du résultat du job sur la
grille de calcul
Comme nous avons déjàmentionné, le
résultat du docking consiste en deux fichiers »dlg (docking log
file) 4 glg (grid log file)», mais le fichier le plus important est
le fichier »dlg».
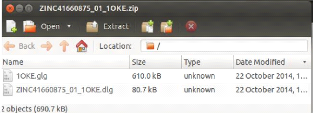
FIGURE 44 - Les fichiers dlg 4 glg du docking
61
La capture ci-après montre que le docking s'est
effectuéavec succès.
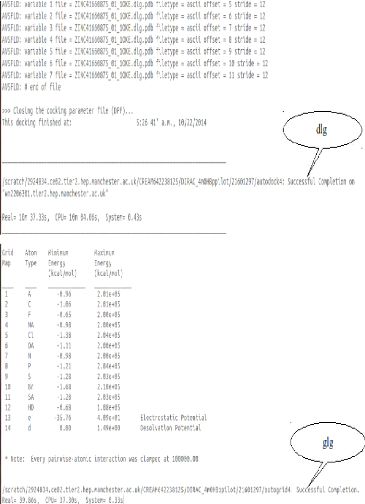
FIGURE 45 - Les fichiers log de docking et de
la grille dlg & glg
Rappelons que le but essentiel de l'utilisation de la grille
de calcul, est la possibilitéde soumettre plusieurs jobs en
parallèle. L'utilisateur peut soumettre plusieurs jobs
de docking sur la grille de calcul via l'intergiciel DIRAC.
Pour cela, il suffit de préparer les fichiers nécessaires pour
réaliser cette opération.
Nous avons préparéles fichiers de
paramètres de docking »dpf» que nous voulons utiliser
afin d'effectuer le docking. Pour soumettre des jobs en parallèle
à l'aide de l'intergiciel DIRAC, nous avons utiliséun workflow.
Ce workflow va générer les
62
fichiers »jdl» essentiels, puis il les
soumets sur la la grille de calcul pour que le Worker Node puisse
exécuter ces jobs. Les résultats des jobs sont
compressédans un fichier zip, ensuite stockésur
l'espace de stockage de la grille. Après que l'opération du
docking s'est terminé, on peut récupérer les
résultat du docking à partir du portail. Afin de montrer la
procédure de docking avec plusieurs fichiers, nous avons préparer
le fichiers de docking »ZINC4.txt», qui comporte les
fichiers ci-dessous :
> ZINC41584388 01 1OKE.dpf >
ZINC41584391 01 1OKE.dpf > ZINC41584955 01 1OKE.dpf >
ZINC41584955 02 1OKE.dpf > ZINC41584983 03
1OKE.dpf
Après avoir soumettre le job, le résultat
consiste en un fichier compresséque nous l'avons récupérer
depuis l'espace de stockage de la grille dès que le docking s'est
achevé. La capture ci-après présente le résultat de
soumission de plusieurs jobs de docking soumis en parallèle.
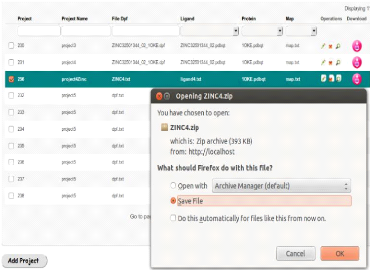
FIGURE 46 - Téléchargement du
résultat des jobs
On accédant à notre espace de stockage de
grille de calcul, on remarque que le résultat du job est bien
stocké. La capture ci-dessous montre le fichiers de paramètre de
docking (ZINC4.txt) et le réusltat (ZINC4.zip).

63
Ce fichier comprend les résultats de tous les jobs
soumet sur la grille de calcul, et chaque fichier contient les deux fichiers
log dlg (docking log file) & glg (grid log file).
FIGURE 47 - Les fichiers des jobs soumis en
parallèle
Mais parfois dûà une mauvaise connexion et
à la non convivialitédes commandes DIRAC, nous ne pouvons pas
récupérer les résultats depuis la grille. Et lors de
récupération des résultats jobs depuis la grille, on s'est
rendu compte que quelques résultats ne sont pas bonnes. Et cela
s'explique par le fait que les données soit de la molécule du
protéine ou celle des ligands contient des informations erronéet
incompatibles.
4.1 Conclusion
Dans le cadre de ce projet, nous avons
utiliséplusieurs outils pour la réalisation de ce projet. Nous
avons utiliséDIRAC comme intergiciel afin de soumettre les jobs
de docking
sur la grille de calcul, suivre l'état du job et
récupérer les résultats. L'outil AutoDock,
oùnous avons déployéla version
AutoDock4.2 pour créer et préparer les fichiers
nécessaires
pour le docking. Et l'outil Taverna pour la
création et la visualisation des workflow. Le but d'utiliser Taverna
est d'avoir la possibilitéd'étendre et d'extensier le
workflow en ajoutant d'autres nouvelles fonctionalités, des services et
d'autres processus pour mieux l'adapter aux besoins ultérieurs des
utilisateurs. Les utilisateurs de ce portail peuvent donc profiter des services
du portail, qui servent comme intermédiare entre les utilisateurs finaux
et les services de la grille. Ce portail fournit un moyen pour la gestion des
protéines, des ligands, des paramètres de grille, des projets de
docking, de soumettre des jobs de docking sur la grille de calcul et de
récupérer les résultats.
64
5 Conclusion & perspective
La découverte de nouveaux médicaments
»in-silico» est l'une des stratégies les plus
prometteuses visant à accélérer le processus de
développement de médicaments. Le criblage virtuel
»Virtual Screening», est l'une des premières
étapes du processus de découverte de médicaments, il
repose sur la sélection »in-silico» des meilleurs
médicaments potentiels qui agissent sur une protéine cible
donnée, il peut se faire »in-vitro», mais il est
très onéreux. Le criblage virtuel nécessite une analyse
complexe avec plusieurs étapes telles que la modélisation
moléculaire et le docking. L'un des principaux avantages
conférés par le docking est qu'il permet aux chercheurs de trier
(screen) rapidement les grandes bases de données de
médicaments potentiels qui nécessiteraient autrement un travail
fastidieux et de longue durée dans le laboratoire selon les
méthodes traditionnelles de découverte de médicaments. La
recherche sur les maladies négligées pourrait largement
bénéficier des avantages de déploiement des grilles
informatiques à plusieurs niveaux. Récemment, le
déploiement de docking »in-silico» sur les grilles de
calcul a émergédans la perspective de réduire les
coûts et le temps de conception de médicaments.
Le présent travail poursuit deux objectifs. Le
premier, est de se familiariser avec l'outil AutoDock afin mieux comprendre le
mécanisme de docking moléculaire protéine-ligand, et
l'outil Taverna pour la création et l'exécution des workflows
scientifiques. Le deuxième objectif consiste à développer
un portail web pour soumettre les jobs de docking in-silico à
grande échelle sur la grille de calcul en utilisant l'intergiciel DIRAC
et l'environnement Taverna [26]. Oùl'utilisateur prépare ses
fichiers nécessaires (protéine, ligand, paramètres de la
grille), soumet son job sur la grille via le portail et récupère
le résultat de son job de docking.
L'achèvement de ce projet implique l'utilisation
coordonnée de plusieurs outils informatiques (AutoDock, Taverna, DIRAC).
Un nombre croissant de ces ressources sont mises à disposition sous la
forme de services Web. De sorte que, ces services Web sont orchestrer dans un
workflow et qui sont mises à la disposition des chercheur scientifiques.
Afin de faciliter l'interaction entre l'utilisateur et les ressources de la
grille de calcul, nous avons développéun portail web qui
répond aux besoins des utilisateurs qui ne sont pas forcément des
experts en informatique. Ce portail permet à ces derniers de charger,
modifier, consulter leur molécules de ligands, protéines sur le
portail. Ainsi, de créer leur projet et d'effectuer le docking
»in-silico» afin d'accélérer leur recherche
sans se préoccuper de la complexitédu portail, tout en
déployant les ressources de la grille de calcul pour soumettre les jobs
de docking à travers l'intergiciel »DIRAC». Ainsi,
nous nous sommes servis de l'outil AutoDock pour préparer les fichiers
nécessaires et effectuer le docking. Nous avons crééun
workflow pour le criblage virtuel sur la grille en utilisant l'outil
»Taverna». Nous
avons pu soumettre les jobs de docking de la base de
donnée ZINC (10256 composés), oùnous avons
stockés les résultats sur l'espace de stockage de la grille de
calcul.
65
Au cours de la réalisation de ce projet, nous avons
rencontréplusieurs des difficultés. Tout d'abord, les
difficultés théoriques. Elles se résument sur la
compréhension des mécanismes de docking moléculaire et de
criblage virtuel. Et les difficultés pratiques se situent au niveau
d'installation et l'utilisation des outils (Taverna, AutoDock). Et au niveau
des lignes de commande de DIRAC, qui ne sont pas assez conviviale à
utiliser. Ainsi, lors de l'utilisation de l'outil Taverna qui consomme
beaucoup de RAM, ce qui entraîne un ralentissement des autres processus
en cours d'exécution et les services nécessaires pour la
soumission des jobs du portail vers la grille de calcul.
Nous allons améliorer le portail web au fur et
à mesure en ajoutant d'autres fonctio-nalités et d'autres
services :
· Mise en place du portail web sur le serveur de l'IFI.
· Authentification au moyen d'un certificat client X509
au lieu du nom et du mot de passe de l'utilisateur.
· Visualisation des résultats de docking
protéine-ligand sous forme de graphe.
Références
[1] Jens Kruger, Richard Grunzke, Sonja Herres-Pawlis,
Performance Studies on Distributed Virtual Screening, 2014
[2] Pratap Parida, Brajesh Shankar, in-silico protein
ligand interaction study of typical antipsychotic drugs against dopaminergic D2
receptor, 2013
[3] William Lindstrom, Garrett M. Morris, Christoph Weber,
Ruth Huey, Using Au-toDock4 for virtual screening, 2008
[4] Romano T. Kroemer, Structure-Based Drug Design
: Docking and Scoring, 2007
[5] C.S.R. Prabhu, Grid and Cluster Computing,
New Delhi : Prentice Hall of India, 2008.
[6] Stian Soiland-Reyes, Ian Dunlop, Alan Williams,
Taverna reloaded
[7] Hsin-Yen Chen, Mason Hsiung, Hurng-Chun Lee, Eric Yen,
Simon C. Lin, Ying-Ta Wu, GVSS : A High Throughput
Drug Discovery Service of Avian Flu and Dengue Fever for EGEE and EUAsiaGrid,
2009
[8] Nguyen Bui The, Nguyen Hong Quang, Doan Trung
Tung,On the Performance Enhancement of the WISDOM Production
Environment, 2012
[9] Ankur Dhanik, John SMcMurray, Lydia Kavraki,
AutoDock based incremental docking protocol to improve docking of large
ligands, 2012
[10] Pritpal Singh, Gurinderpal Singh, Grid computing
architecture. 2013
[11] JACQ Nicolas, In silico drug discovery services
in computing grid envir nments against neglected and emerging infectious
diseases. 2006
[12] D. Hull, K. Wolstencroft, R. Stevens, C. A. Goble, M. R.
Pocock, Taverna: a tool for building and running workflows of services,
Nucleic Acids Research , 34 (Web-Server-Issue) :729 732, 2006
[13] Quang Bui The, Doan Trung Tung, Nguyen Hong Quang,
Criblage virtuel sur grille de composés isolés au
Vietnam, 2011
[14] Yasmine Asses, Conception par modélisation
et criblage in silico d'hinibi-teurs du récepteur c-Met,
2011
[15] Grosdidier A.Conception d'un logiciel de docking et
applications dans la recherche de nouvelles molécules actives,
Thèse de doctorat en pharmacie. Grenoble :
UniversitéJoseph Fourier. France. 2007
[16] Sousa S. F., Fernandes P. A., Ramos M. J,
Protein-Ligand Docking: Current Status and Future Challenges,
Proteins. 2006; 65 : 15-26
[17] Koblitz, B.Santos, N.Pose, The AMGA metadata
service, J.Grid Computing6,61-67 (2008)
[18] German-Renaud, C.Loomis, Scheduling for
responsive grids, J.Grid Computing 7,463-478 (2009)
[19] A.Tsaregorodtsev, V.Hamar, M.Sapunov, T.Glatard,
Infrastructure DIRAC pour les Communautés Pluridisciplinaires,
2011
[20] A.Tsaregorodtsev, P. Charpentier, V.Romanovski,
DIRAC - The Distributed MC Production and Analysis for LHCb ,
2010
[21] E. van Herwijnen, J. Closier, M. Frank, C. Gaspar, F.
Loverre, S.Ponce, and M. Gan-delman, Dirac-distributed infrastructure
with remote agent control, Conference for Computing in High-Energy and
Nuclear Physics (CHEP 03), 2003.
[22] Bui The Quang, Nguyen Hong Quang, Emmanuel Medernach,
Vincent Breton , Multi-Level Queue-Based Scheduling for Virtual
Screening Application on Pilot-Agent Platforms on Grid/Cloud to Optimize the
Stretch , 2014
[23] Katherine Wolstencroft, Robert Haines, Donal Fellows,
Alan Williams, The Taverna workflow suite : designing and executing
workflows of Web Services on the desktop, web or in the cloud, 2013
[24] Trung Tung DOAN, Quang Minh DAO, Trong Hieu VU, Hong
Phong PHAM, g-INFO portal: a solution to monitor Influenza A on the
Grid for non-grid users, 2011
[25] Nicolas JACQ, Jean SALZEMANNa, Yannick LEGRE, Matthieu
REICHS-TADT,Demonstration of In Silico Docking at a Large Scale on Grid
Infrastructure , 2006
[26] T. Oinn, M. Addis, J. Ferris, D. Marvin, M. Senger, M.
Greenwood, T. Carver, K. Glover, M. R. Pocock, A.Wipat, P. Li, Taverna
: A tool for the composition and enactment of bioinformatics
workflows, Bioinformatics Journal 20(17) pp 3045-3054, 2004, doi
:10.1093/bioinformatics/bth361.
I
Annexe
· Installation du client DIRAC :
Avant d'installer DIRAC, l'utilisateur doit respecter les
conditions suivantes :
- Doit être un membre d'une organisation
virtuelle par exemple : euasia,
biomed,...
Nous nous sommes enregistréauprès de la VO Biomed
via le site :
https: //
cclcgvomsli01. in2p3.
fr: 8443/ voms/ biomed/ user/ home. action
- Possède un certificat X.509 reconnu
par EGEE, afin de pouvoir utiliser les ressource de la grille EGEE.
· II
Script pour préparer les fichiers de grille et de docking
»gpf et dpf»
#!/bin/sh
WORK DIR=`pwd`
MGTOOLS="/usr/local/MGLTools-1.5.6/MGLToolsPckgs/AutoDockTools/
Utilities24"
PYTHON="/usr/local/MGLTools-1.5.6/bin/pythonsh"
while getopts "l:r:" opt; do
case $opt in
l)
LIG FILE=$OPTARG
LIG BASE NAME=$(basename $LIG FILE)
LIG EXT=${LIG FILE##*.}
[ -f "${OPTARG}" ] && if [ "$LIG EXT" = "pdbqt" ];
then echo "Ligand file name " "$LIG FILE"
else echo "Check the ligand file and extension"
fi ;;
r)
PROT FILE=${OPTARG}
PROT BASE NAME=$(basename "$PROT FILE")
PROT EXT=${PROT BASE NAME##*.}
echo "Extension = $PROT EXT"
[ -f "$PROT FILE" ] && if [ "$PROT EXT" = "pdbqt"
];
then
echo "Protein file name " "$PROT FILE"
else
echo "Check the protein file and extension"
fi
;;
*)
echo "Require argument!!"
exit 1
;;
esac
done
shift $((OPTIND-1))
ulimit -s unlimited
cd $WORK DIR
$PYTHON prepare
gpf4.py -l $LIG FILE -r $PROT FILE -o
ResGrid.gpf
$PYTHON prepare
dpf4.py -l $LIG FILE -r $PROT FILE -o
ResDock.dpf
· III
Liste des jobs de docking soumis à travers le portail sur
la grille de calcul et leur états.

·
IV
Résultats des jobs de docking stockés sur l'espace
de stockage de la grille de calcul.

| 

