
2009-2010
Département Informatique
MEMOIRE DE MAGISTER
Option : Informatique et Automatique
THEME :
FOUILLE DE DONNEES BIOLOGIQUES : ETUDE COMPARATIVE
ET
EXPERIMENTATION.
Présenté par : Abdelhak
MANSOUL
Soutenu devant les membres du jury :
Mr B. BELDJILALI Professeur à l'Université
d'Oran
Président
Mr K. BOUAMRANE Maître de Conférences à
l'Université d'Oran
Examinateur
Mr A. GHOMARI Maître de conférences à
l'Université d'Oran
Examinateur
Mr M. MALKI Maître de conférences à l'UDL
de Sidi Belabess
Examinateur
Mr B. ATMANI Maître de conférences à
l'Université d'Oran
Rapporteur
Résumé
Le traitement des données biologiques est
indispensable en recherches médicales et sciences de la vie. En effet,
les données biologiques sont de différents types, et souvent
complexes, ce qui a induit une recherche soutenue de nouveaux
procédés d'exploitation parce que ceux existant ne suffisent plus
ou ne sont plus adaptés. Une nouvelle approche : l'Extraction de
Connaissances à partir des Données biologiques est de plus en
plus envisagée. De là, notre étude qui porte sur la
fouille de données biologiques sur un terrain expérimental : une
épidémie.
Le présent travail de recherche se situe dans
le cadre de l'ECD Biologiques, à travers une étude comparatives
des outils existants et la proposition d'une nouvelle approche pour
l'extraction des règles d'association à partir de données
biologiques, leur gestion et l'alimentation d'un système d'aide à
la décision.
D'où, la problématique
abordée par notre étude qui est la fouille de données
biologiques assistée par une modélisation booléenne des
résultats obtenus.
Nous proposons un processus d'extraction de motifs
assez novateur pour générer des règles d'association
profitable et exploitable à deux niveaux :
· Profitable au spécialiste du domaine,
en particulier à travers les règles d'association qui aident
à mieux interpréter les données.
· Le résultat de la fouille de
données est optimisé par une modélisation booléenne
des règles d'association extraites. Cette amélioration se fait
par la machine BRI (Boolean Rules Induction ). En premier lieu nous
présenterons un état de l'art, s'ensuit une étude
comparative des différents outils et méthodes existants afin d'en
tirer bénéfice, et on continuera par exposer notre
démarche et les résultats obtenus.
Mots clés: Automate cellulaire, Fouille de
données biologiques, Induction de règles, Règle
d'association, modélisation booléenne.
Abstract
The biological data processing is an
indispensable tool in medical researches and life sciences. Indeed, the
biological data are various types, and often complex, what led a search of new
exploitation processes because those existing are not any more enough or are
not any more adapted. A new approach : the Extraction of Knowledge from the
biological data is more and more envisaged. From there, our study which
concerns the Biological Data Mining on an experimental ground: an
epidemic.
The present research work is situated within the
framework of Knowledge Discovery from Biological Data, through study
comparative clauses of the existing tools and the proposition of a new approach
for the extraction of the association rules from biological data, there
management and the supply of a system of decision-making
support.
Where from, the problem approached by our study
which is the Data Mining of biological data assisted by a boolean modeling for
the obtained results.
We propose a rather innovative process of extraction
of patterns for generating a profitable and
exploitable association rules at two
levels:
· Profitable, to the specialist of the domain,
in particular through the rules of association which help to interpret better
the data.
· The result of the data mining process is
optimized by a boolean modelling of the extracted association rules. This
improvement is made by the machine BRI (Boolean Rules Rules
Induction).
First of all we shall present a state of the art,
follows a comparative study of the various existing tools and the methods to
benefit from it, and we shall continue to expose our approach and the obtained
results.
Key words: Cellular automaton, Biological data
mining, Rules Induction, Association Rules, Boolean
modelisation
Remerciements
Je remercie les membres du jury qui m'ont fait
l'honneur d'avoir accepté d'évaluer ce travail.
Je remercie vivement Monsieur Bouziane BELDJILALI,
qui m'a bien accueilli et m'a entretenu pour me diriger ensuite vers mon
encadreur. Ainsi que, Monsieur Baghdad ATMANI mon encadreur, pour m'avoir
dirigé pendant tout le long de ce travail, par ses précieux
conseils, ses pertinents commentaires, et ses orientations. De plus m'a fait
profiter de son expérience dans la direction de travaux de
recherche.
Mes remerciements vont aussi :
À Monsieur Abdelhafid HAFFAF, le chef du
département informatique de l'université d'Oran.
À Monsieur Karim BOUAMRANE pour m'avoir
facilité les démarches administratives au département
informatique.
Et Monsieur Smain MAAZOUZI, le chef du
département informatique de l'université du 20 Août 55 de
SKIKDA pour son grand soutien.
TABLE DES MATIERES
Résumé
Liste des figures
Liste des tableaux
Glossaire
Introduction générale 1
Chapitre I. L'Extraction de Connaissances à partir
de Données Biologiques 6
I.1 Définition de l'extraction de connaissances à
partir de données
biologiques 6
I.2 Le processus de l'ECD biologiques 7
I.3 Notre contribution 13
I.4 Etat de l'art de l'ECD biologiques 14
I.5 Les méthodes de fouille de données 20
I.6 Etude comparative 27
I.7 Discussion sur l'ECD Biologiques 30
I.8 Conclusion 31
Chapitre II. Extraction de règles d'association
33
II.1 Les règles d'association 34
II.2 L'induction et l'évaluation des règles 35
II.3 Les algorithmes d'extraction des règles d'association
37
II.4 Conclusion 42
Chapitre III. Modélisation booléenne des
règles d'association 44
III.1 Le moteur d'inférence cellulaire : architecture et
principe de 44
fonctionnement
III.2 La modélisation booléenne 47
III.3 Exemple d'illustration d'induction des règles
booléennes 48
III.4 La dynamique du moteur d'inférence cellulaire 50
III.5 Conclusion 52
Chapitre IV. Conception et expérimentation du
système BIODM 54
IV.1 Etude et choix des données biologiques pour
expérimentation 54
IV.2 Architecture du système BIODM (BIOlogical Data
Mining) 55
IV.3 Le processus de l'ECD biologiques 57
IV.4 Le logiciel réalisé 63
IV.5 L'expérimentation 66
IV.6 Conclusion 70
Conclusion générale 71
Références bibliographiques 73
Annexe B 77
Liste des figures
Introduction générale.
Figure 0.1 : Complexe Tuberculosis. 2
Figure 0.2 : Morceau de séquence génomique
rapatriée de NCBI. 4
Figure 0.3 : Fichier des séquences ayant subi une
transformation. 4
Chapitre I. L'ECD Biologique.
Figure 1.1 : Exemple du format FASTA d'une séquence
protéique. 9
Figure 1.2 : Exemple du format STADEN d'une séquence
protéique. 9
Figure 1.3 : Exemple du format PIR d'une séquence
protéique. 10
Figure 1.4 : Exemple de fichier à l'état brut de la
séquence génomique de la
souche MT CDC1551 au format texte brut. 10
Figure 1.5 : Morceau de la séquence génomique
nettoyée du Mt CDC1551. 11
Figure 1.6 : Morceau de la séquence génomique mise
en forme du
Mt CDC1551. 11
Figure 1.7 : Morceau de la séquence génomique
structurée du Mt CDC1551. 11
Figure 1.8 : Processus d'ECD Biologiques. 12
Chapitre III. Modélisation booléenne des
règles d'association.
Figure 3.1 : Le système BRI (Boolean Rule Induction).
44
Figure 3.2 : Les partitions S, S1 et S2. 45
Figure 3.3 : Illustration du principe d'induction des
règles booléennes
inductives par BRI. 48
Chapitre IV. Conception et expérimentation du
système BIODM.
Figure 4.1 : Architecture du système BIODM. 55
Figure 4.2 : Morceaux de la séquence génomique du
Mt CDC1551. 58
Figure 4.3 : Morceaux de séquence protéique du Mt
CDC1551. 58
Figure 4.4 : Architecture fonctionnelle du système BIODM.
64
Figure 4.5 : Interface du système BIODM. 66
Figure 4.6 : Echantillon de gènes servant à la
fouille de données. 67
Liste des tableaux
Introduction générale.
Tableau 0.1: Tableau des différentes souches du
Mycobacterium Tuberculosis 77
Tableau 0.2: Tableaux informatif sur les
caractéristiques des souches du
Mycobacterium Tuberculosis complètement annotées.
78
Tableau 1.3 : Les souches du Mycobacterium Tuberculosis en cours
d'annotation. 78
Chapitre I. L'ECD Biologique.
Tableau 1.1: Description du fichier FASTA de l'exemple de la
figure 1.1. 9
Tableau 1.2: Description du fichier PIR de l'exemple de la figure
1.3. 10
Tableau 1.3: Les souches du Mycobacterium Tuberculosis en cours
d'annotation. 77
Tableau 1.4: Les méthodes de FDD utilisées en ECD
biologiques. 28
Tableau 1.5: Les tâches et méthodes utilisées
en ECD. 29
Tableau 1.6: Tableau comparatif des tâches de l'ECD. 29
Chapitre III. Modélisation booléenne des
règles d'association
Tableau 3.1 : Représentation cellulaire de la Base des
connaissances de la
figure 3.2. 46
Tableau 3.2 : Les matrices d'incidences
d'entrée RE et de sortie Rs pour la
figure 3.2. 46
Chapitre IV. Conception et expérimentation du
système BIODM.
Tableau 4.1 : Base de test servant à
l'expérimentation. 66
Tableau 4.2 : Exemple de règles
générées par Apriori pour un support de 60% 68
et une confiance de 80%.
Tableau 4.3 : Exemple de règles cellulaires
générées par BRI. 68
Tableau 4.4 : Nombre de
règles et temps d'exécution d'Apriori sur l'échantillon
de la figure 4.6. 69
Tableau 4.5 : Evolution de l'espace de stockage. 69
Annexe A
Glossaire
A
Acide désoxyribonucléique
(ADN)
Support biochimique de l'information génétique
chez tous les êtres vivants (à l'exception de quelques virus qui
utilisent l'ARN). Principal composant des chromosomes, l'ADN se présente
le plus souvent sous forme de deux longs filaments (ou chaînes)
torsadés l'un dans l'autre pour former une structure en double
hélice. Chacune de ces chaînes est un polymère formé
de l'assemblage de quatre nucléotides différents,
désignés par l'initiale de la base azotée qui entre dans
leur composition : A (Adénine), C (Cytosine), G (Guanine) et T
(Thymine).
Acide ribonucléique (ARN)
Dans les cellules, on distingue plusieurs types d'ARN suivant
leur fonction. Les trois types principaux sont : les ARN messagers, les ARN de
transfert et les ARN ribosomaux. L'ARN est un acide nucléique
constitué d'une seule chaîne de nucléotides, de structure
analogue à celle de l'ADN. Il existe cependant des différences
chimiques entre ces deux acides nucléiques qui donnent à l'ARN
certaines propriétés particulières. L'ARN est produit par
transcription de l'ADN.
ACP
L'analyse en composantes principales (ACP) est une
méthode mathématique d'analyse des données qui consiste
à rechercher les directions de l'espace qui représentent le mieux
les corrélations entre n variables aléatoires
Acyclique (graphe)
Un graphe acyclique est un graphe ne contenant aucun cycle.
Agrégation (données)
Le mot agrégation désigne l'action
d'agréger, de regrouper des éléments.
Alignement Global / Local
L'alignement de séquences (ou alignement
séquentiel) est une manière de disposer les composantes
nucléotides ou acides aminés) des ADN, des ARN, ou des
séquences primaires de protéines pour identifier les zones de
concordance qui traduisent des similarités ou dissemblances de nature
historique. Il existe l'alignement global, c'est-à-dire entre les deux
séquences sur toute leur longueur (FASTA) et local, entre une
séquence et une partie de l'autre séquence (BLAST).
Annotation
L'annotation d'un génome consiste à traiter
l'information brute contenue dans la séquence dans le but :
1. de prédire, le contenu en gènes, la position
des gènes à l'intérieur d'un génome ainsi que leur
organisation, des séquences promotrices, etc. Dans ce cas, on parle
d'annotation structurale.
2. de prédire la fonction potentielle de ces
gènes. Dans ce cas on parle d'annotation fonctionnelle.
Antigènes
Un antigène est une macromolécule naturelle ou
synthétique, reconnue par des anticorps ou des cellules du
système immunitaire et capable d'engendrer une réponse
immunitaire.
Arbre de décision
Modèle issu des techniques d'intelligence artificielle.
Son principe est de chercher à diviser une population en 2 (arbres
binaires) ou plus (arbres n-aires) de sorte que ces sous-populations soient
aussi différentes entre elles que possibles, et homogènes du
point de vue de la répartition de la variable cible.
Apprentissage (échantillon
d')
Partie des données servant à l'évaluation
des différents paramètres d'un modèle (en anglais,
"training").
Athérosclérose
Le vieillissement normal des artères et artérioles
se nomme artériosclérose.
Auto-immunes (maladies)
Les maladies auto-immunes sont dues à une
hyperactivité du système immunitaire à l'encontre de
substances ou de tissus qui sont normalement présents dans
l'organisme.
Automate cellulaire
Un automate cellulaire consiste en une grille
régulière de « cellules » contenant chacune un «
état » choisi parmi un ensemble fini et qui peut évoluer au
cours du temps. L'état d'une cellule au temps t+1 est fonction de
l'état au temps t d'un nombre fini de cellules appelé son «
voisinage ». À chaque nouvelle unité de temps, les
mêmes règles sont appliquées simultanément à
toutes les cellules de la grille, produisant une nouvelle «
génération » de cellules dépendant entièrement
de la génération précédente.
Annexe A
B
Bio-informatique
La Bio-informatique est constituée par l'ensemble des
concepts et des techniques nécessaires à l'interprétation
de l'information génétique (séquences) et structurale.
C'est le décryptage de la « bio-information ». La
bio-informatique est donc une branche théorique de la biologie.
Biologie moléculaire
La biologie moléculaire est une discipline scientifique au
croisement de la génétique, de la biochimie et de la physique,
dont l'objet est la compréhension des mécanismes de
fonctionnement de la cellule au niveau moléculaire.
BLAST
BLAST (acronyme de basic local alignment search tool) est une
méthode de recherche heuristique utilisée en bio-informatique
permettant de trouver les régions similaires entre deux ou plusieurs
séquences de nucléotides ou d'acides aminés.
C
Candidat (gène)
L'approche gène candidat consiste à supposer
l'implication d'un gène dans un quelconque effet à priori, et
l'étude vise à confirmer cette implication a posteriori.
Cas-témoins (étude)
Etude rétrospective entre deux groupes, l'un
présentant une maladie (cas) et l'autre, indemne (témoins).
Chromosome
Unité physique de matériel
génétique correspondant à une molécule continue
d'ADN. Les cellules bactériennes n'en comportent qu'un. Ils sont
doués du pouvoir d'autoreproduction.
Classification ascendante hiérarchique
(CAH)
Méthode de création de typologies qui
agrège, à chaque étape, les individus ou les groupes
d'individus les plus proches. Les emboîtements successifs se poursuivent
ainsi jusqu'à agréger toute la population. On choisit ensuite la
partition (ensemble de classes ainsi constituées) qui propose le
meilleur rapport homogénéité interne des groupes /
hétérogénéité des groupes entre eux.
Classification automatique
On appelle classification automatique la catégorisation
algorithmique d'objets. Celle-ci consiste à attribuer une classe ou
catégorie à chaque objet (ou individu) à classer, en se
basant sur des données statistiques.
Coeliaque (maladie)
La maladie coeliaque est une maladie auto-immune,
caractérisée par une atteinte de tout ou partie des
villosités recouvrant l'intestin grêle.
Co-régulé
(gène)
Gènes liés l'un à l'autre.
Code génétique
Système de correspondance permettant de traduire une
séquence d'acide nucléique en protéine.
Cohorte
Ensemble d'individus étudiés sur une
période de temps donnée. Une cohorte permet de suivre de
manière longitudinale les comportements de la population observée
ainsi que sa réaction à un ou plusieurs événements
donnés.
Continue (variable)
Se dit d'une variable qui peut prendre une "infinité"
de valeurs (par opposition à discrète) par exemple, un
réel. Un âge, une somme d'argent, un coefficient de bonus/malus
sont souvent considérés comme continus. Synonyme :
quantitatif.
Corrélation
Mesure de la liaison entre deux variables. On parle de
corrélation entre une cause et son effet, ou entre deux variables qui
apportent la même information.
CROHN (maladie)
La maladie de Crohn est une maladie inflammatoire chronique
intestinale (MICI) de l'ensemble du tube digestif.
Annexe A
D
Data Mining (outils de)
Aussi connu sous le nom de KDD (Knowledge Discovery Data), les
outils de data mining permettent d'extraire de la connaissance des
données en découvrant des modèles, des règles dans
le volume d'information.
Data mining
Le terme anglais «datamining» évoque le
travail de «mineur de fond» pour extraire les données
pertinentes noyées dans de gros volumes de données. Ensemble de
techniques héritées de la statistique "classique", de la
statistique bayésienne et de l'intelligence artificielle, qui permet
l'étude de grands volumes de données. Ces techniques sont
soutenues en général par une méthode de travail qui pose
les étapes de l'étude DataMining.
Déduction / induction
En logique, la déduction procède de la
conception que les moyens ne sont pas plus importants que la fin (conclusion),
par opposition à l'induction logique qui consiste à former des
représentations générales à partir de faits
particuliers.
Dichotomique (Variable)
Variable qui peut opérer une division de
l'échantillon en deux parties.
Discrète / Continue
(variable)
Se dit d'une variable qui ne prend qu'un nombre limité
et connu d'avance de modalités (valeurs distinctes), par opposition
à continue. Une situation familiale, un sexe, ou à une
catégorie socio-professionnelle sont des variables discrètes.
Synonyme : qualitative.
Distance
En mathématiques, une distance est une application qui
formalise l'idée intuitive de distance, c'est-à-dire la longueur
qui sépare deux points.
Données biologiques ( cohorte
)
Ce sont les des dosages systématiques
réalisés (la biochimie, NFS numération de formule sanguine
et analyse d'urine).
Données cliniques ( cohorte )
Les données cliniques, se divisent en examens cliniques
systématiques (taille, poids, pression artérielle, ....), et en
examens cliniques spécifiques (échographie,.....).
Données
génétiques
Les données relatives au génome (ADN, ..).
E
Élaguer
Consiste à supprimer d'un problème des valeurs de
variables ne pouvant pas prendre part à une solution.
Épi-génétique
(maladie)
Le terme épigénétique définit les
modifications transmissibles et réversibles de l'expression des
gènes ne s'accompagnant pas de changements des séquences
nucléotidiques.
Epidémiologie
Etude des différents facteurs qui interviennent dans
l'apparition et l'évolution des maladies.
Eucaryotes / procaryotes
L'ensemble des organismes vivants peut être
classé en trois grands groupes : les eucaryotes (L'Homme, ainsi que les
animaux, les plantes et les champignons), les eubactéries, les
archaebactéries. Les cellules des eucaryotes possèdent un noyau.
Les eubactéries et les archaebactéries ne possèdent pas de
vrai noyau.
F
FASTA
C'est une méthode de recherche heuristique
utilisée en bio-informatique permettant de trouver les régions
similaires entre deux ou plusieurs séquences de nucléotides ou
d'acides aminés. Ce programme permet de retrouver rapidement dans des
bases de données, les séquences ayant des zones de similitude
avec une séquence donnée (introduite par l'utilisateur).
Annexe A
Fonctionnelle (génomique)
Étude de la fonction des gènes par analyse de leur
séquence et de leurs produits d'expression : les ARNm (transcriptome) et
les protéines (protéome).
G
Gène
Fragment d'ADN portant les informations nécessaires
à la fabrication d'une ou plusieurs protéine(s). Un gène
comprend la séquence en nucléotide qui peut varier de quelques
centaines, à plus d'un million de nucléotides.
Génétique (algorithme)
Un algorithme génétique est un algorithme lent,
représentant les modèles comme des gènes et des
opérateurs génétiques et les faisant évoluer soit
par mutation (un gène au hasard est remplacé), soit par
cross-over (la place de deux sous-arbres est échangée).
Génome
Ensemble de l'information génétique d'un organisme
(matériel génétique présent dans chacune des
cellules d'un individu, patrimoine héréditaire d'un individu).
Une copie du génome est présente dans chacune de ses cellules. Le
génome est transmis de génération en
génération.
Génomique
Étude des génomes. Son objectif est de
séquencer l'ADN d'un organisme et de localiser sur celui-ci tous les
gènes qu'il porte, puis de caractériser leurs fonctions.
Génotype
Ensemble des caractères génétiques d'un
individu. Son expression conduit au phénotype.
H
HMM
Un modèle de Markov caché (MMC) -- en anglais
Hidden Markov Models (HMM) (ou plus correctement, mais moins employé
automate de Markov à états cachés) est un modèle
statistique dans lequel le système modélisé est
supposé être un processus Markovien de paramètres inconnus.
Les modèles de Markov cachés sont massivement utilisés
notamment en reconnaissance de formes, en intelligence artificielle ou encore
en traitement automatique du langage naturel.
I
Induction
Méthode consistant à tirer une conclusion d'une
série de faits. Cette conclusion ne sera jamais sûre à 100
%. L'induction en revanche génère du sens en passant des faits
à la loi, du particulier au général.
M
Marqueur génétique
En cartographie génétique, séquence d'ADN
particulière utilisée pour "baliser" les chromosomes.
Modèle
Mécanique plus ou moins "boîte noire" qui,
à partir de données connues (input), calcule une réponse
(target) et la probabilité de réalisation de cette réponse
associée (score).
Moteur d'inférence
Partie d'un système expert qui effectue la
sélection et l'application des règles en vue de la
résolution d'un problème donné.
Motifs fréquents
Un caractère ou trait qui se répète
fréquemment.
Motifs séquentiels
Les motifs séquentiels permettent de traiter de gros
volumes de données et d'en extraire des règles incluant la
dimension temporelle
Mutation
Modification affectant l'ADN d'un gène. Cette
altération du matériel génétique d'une cellule ou
d'un virus entraîne une modification durable de certains
caractères du fait de la transmission héréditaire de ce
matériel de génération en génération.
Annexe A
N
Nucléotide
Motif structural de base des acides nucléiques,
formé de l'assemblage de plusieurs molécules : un sucre, un acide
phosphorique et une base azotée (dans le cas de l'ARN, cette base peut
être l'Adénine - A, la Cytosine - C, la Guanine - G ou l'Uracile -
U ; idem dans le cas de l'ADN, excepté que l'Uracile est remplacé
par la Thymine - T).
O
OR (Odds Ratio)
Un Odds ratio (OR), se définit comme le rapport des
chances qu'un évènement arrivant, par exemple une maladie,
à un groupe de personnes A, arrive également à un autre
groupe B.
Orphelines (pathologies)
Les maladies rares ou maladies orphelines sont des maladies qui
affectent moins de 0,05 % de la population (1 personne sur 2 000).
P
Pathogènes
/pathogénicité
Les agents infectieux sont un type d'agent pathogène,
responsables des maladies infectieuses.
PE / PPE
Familles de protéines.
Perceptron
Catégorie de réseaux de neurones robustes. Ils
diffèrent des autres réseaux (les RBF) par la fonction
d'activation des neurones, c'est à dire leur manière de
transformer les signaux d'entrée en signal de réponse.
Plasmide
Petite molécule circulaire d'ADN extrachromosomique
présente chez les bactéries, capable de se répliquer de
façon autonome, dans la cellule d'origine et dans une
cellule-hôte.
Polymorphismes
génétiques
Les polymorphismes génétiques s'expriment chez les
individus sous la forme de différents phénotypes.
Protéine
L'un des quatre matériaux de base de tout organisme, avec
les glucides, les lipides et les acides nucléiques. Les protéines
sont formées d'un enchaînement spécifique d'acides
aminés (de quelques dizaines à plusieurs centaines). Les
protéines remplissent différentes fonctions dans la cellule,
notamment des fonctions de structure et des fonctions enzymatiques.
Protéome / protéomique
Le protéome est l'ensemble des protéines produites
à partir du génome d'un organisme. La protéomique est
l'étude du protéome, dans le but de déterminer
l'activité, la fonction et les interactions des protéines.
Puce à ADN
Technologie employée dans l'étude du transcriptome
et basée sur la capacité des molécules d'ADN et d'ARN
à s'hybrider entre elles. De courtes séquences d'ADN connues sont
fixées sur des supports d'une surface de l'ordre du centimètre
carré : les puces.
Q
Qualitative / Quantitative
(variable)
Une variable qualitative est une variable pour laquelle la
valeur mesurée sur chaque individu (parfois qualifiée de
catégorie ou de modalité) ne représente pas une
quantité. Une variable est dite quantitative lorsque la valeur
mesurée sur chaque individu représente une quantité.
R
Raisonnement à partir de cas / Case Based
Reasoning
Un système CBR dispose d'une base de cas. Chaque cas
possède une description et une solution. Pour utiliser ces informations,
un moteur est aussi présent. Celui-ci va retrouver les cas similaires au
problème posé. Après analyse, le moteur fournit une
solution adaptée qui doit être validée. Enfin le moteur
ajoute le problème et sa solution dans la base de cas.
Annexe A
Règles séquentielles
C'est une règle d'association incluant le facteur
temporel.
Renforcement (apprentissage)
L'apprentissage par renforcement fait référence
à une classe de problèmes d'apprentissage automatique, dont le
but est d'apprendre, à partir d'expériences, ce qu'il convient de
faire en différentes situations, de façon à optimiser une
récompense numérique au cours du temps.
RR (Risque relatif),
Le risque relatif (RR) est une mesure statistique souvent
utilisée en épidémiologie, mesurant le risque de survenue
d'un événement entre deux groupes.
S
Segmentation (ou Typologie)
Découpage d'une population en fonction d'un ou
plusieurs critères (géographiques, sociodémographiques,
comportementaux...). Les groupes ainsi constitués aussi homogènes
et différents entre eux que possibles, peuvent être choisis comme
autant de cibles à atteindre à l'aide d'un marketing mix
spécifique.
Séquençage
(génome)
Analyse du génome, consistant à
déterminer la succession de toutes les bases qui composent l'ADN d'un
organisme. Ce séquençage n'est réalisé ou en cours
de réalisation que pour un nombre limité d'espèces :
quelques bactéries, une levure, un insecte (la drosophile) et l'homme.
Le séquençage ne permet pas la détermination de la
fonction des protéines codées par l'ADN.
Séquenceurs automatiques
Un séquenceur de gènes (ou « séquenceur
») est un appareil capable d'automatiser l'opération de
séquençage de l'ADN.
Séquences répétées
directes
Séquences identiques ou quasi identiques, présentes
en plusieurs copies dans la même molécule d'ADN.
Séquences répétées en
tandem
Séquences répétées directes
adjacentes.
Souche (bactérie)
Une population d'une espèce pouvant engendrée
une population fille c'est-à-dire les ancêtres d'une population,
par exemple des souches de bactéries pathogènes,
Supervisé / non supervisé
(méthode)
L'apprentissage supervisé est une technique
d'apprentissage automatique où l'on cherche à produire
automatiquement des règles à partir d'une base de données
d'apprentissage contenant des exemples de cas déjà
traités. L'apprentissage non-supervisé est une méthode
d'apprentissage automatique. Cette méthode se distingue de
l'apprentissage supervisé par le fait qu'il n'y a pas de sortie a
priori.
Streptococcus
Les Streptococcus ou streptocoques sont des bactéries.
On retrouve des streptocoques un petit peu partout dans la nature. Certains
vivent sur la peau et les muqueuses de l'homme : leur présence est
normale.
Syndrome métabolique
Le syndrome métabolique (ou syndrome X)
désigné par les acronymes SMet (pour syndrome métabolique)
ou MetS (pour Metabolic syndrome chez les anglophones) désigne
l'association d'une série de problèmes de santé ayant en
commun un mauvais métabolisme corporel.
T
Transfert horizontal / vertical
Le Transfert horizontal de gènes (ou HGT pour
Horizontal Gene Tranfer en anglais), est un processus dans lequel un organisme
intègre du matériel génétique provenant d'un autre
organisme sans en être le descendant. Par opposition, le transfert
vertical se produit lorsque l'organisme reçoit du matériel
génétique à partir de son ancêtre.
Introduction générale - 1 -
Introduction générale
Au cours des dernières années, la
bioinformatique [Gibas et Jambeck, 2002] a connu un grand développement
lié à l'aboutissement de nombreux travaux de
séquençage, lesquels ayant conduit à l'arrivée
d'énormes quantités de données biologiques qu'il faut
exploiter pour tirer un maximum de connaissances possibles [Chervitz et al.,
1999], [Tzanis et al., 2005]. Si dans un premier temps, les génomes
séquencés étaient ceux des procaryotes (unicellulaires :
Bactérie, ....), nous arrivons maintenant au stade où des
génomes d'eucaryotes (pluricellulaires : animaux, humains, ...) sont
disponibles. De ce fait, les quantités de données brutes
disponibles sont déjà trop importantes pour pouvoir être
analysées manuellement [Chervitz et al., 1999].
L'outil informatique et par la même les
méthodes informatiques se sont imposées d'elles mêmes en
biologie moléculaire: C'est la naissance de la bioinformatique. Son
développement a été rendu possible par les énormes
progrès réalisés en informatique (capacités de
calcul, stockage, nouveaux algorithmes,...), qui ont permis la constitution de
banques de données pour le stockage de l'intégralité des
données biologiques produites par les
expérimentations.
Dans un autre volet complémentaire, nous avons
l'épidémiologie, qui est basée sur l'utilisation des
méthodes de surveillance et d'analyse des données recueillies
concernant les diagnostics relatifs à des infections. Ces
méthodes classiques ne sont plus satisfaisantes comme elles
l'étaient autrefois, surtout quand il s'agit d'analyser et
détecter précocement une épidémie causée par
une maladie émergente.
Du fait de l'inefficacité de ces
méthodes, de la variété des données biologiques, et
de la nature même des épidémies [Labbe, 2007], une nouvelle
approche, exploitant des données biologiques relatives aux
épidémies, est utilisée afin de mieux comprendre les
maladies qui ont un profil épidémiologique : c'est la fouille de
données biologiques relatives aux épidémies [Remvikos,
2004], [Maumus et al., 2005], [Etienne, 2004].
Cette fouille de données permet d'extraire des
connaissances qui aideront à mieux connaître ou interpréter
les phénomènes biologiques liés à une
épidémie particulière et ainsi permettre la mise en oeuvre
de mesures de prévention et de lutte, par des traitements
appropriés, des vaccinations, des antibiotiques, etc.
Un autre aspect, la disponibilité de vastes
banques de données de santé publique relatives aux
épidémies issues des récents séquençages de
nouveaux agents pathogènes,
Introduction générale - 2 -
a incité à les valoriser pour mieux
connaitre les épidémies et aider les spécialistes à
trouver des réponses thérapeutiques efficaces.
En effet, parmi ces épidémies, il existe
une qui a montré un fort intérêt notamment par les
récents séquençages de nouvelles souches : c'est la
tuberculose.
A l'origine l'infection est provoquée par la
pénétration dans l'organisme d'une bactérie appelée
Mycobacterium Tuberculosis, et lorsque cette infection se multiplie dans un
lieu et une période donnée cela abouti à une
l'épidémie. Dans la pratique, il existe un Complexe Tuberculosis
dont le Mycobacterium Tuberculosis est l'agent typique responsable de la
tuberculose humaine (voir Figure 0.1).
Complexe Tuberculosis

M.
Tuberculosis
M.
Africanum
M.
Bovis
M. Bovis BCG
M.
Canetti
M.
Microti
Figure 0.1: Complexe Tuberculosis.
L'agent pathogène : La bactérie
Les bactéries (Bacteria) sont des organismes
vivants unicellulaires. Elles mesurent quelques micromètres de long et
peuvent présenter différentes formes : des formes
sphériques (coques), allongées ou en bâtonnets (bacilles),
et des formes plus ou moins spiralées [Wikipedia].
Caractéristiques génétiques d'une
bactérie
La plupart des bactéries possèdent un
unique chromosome circulaire, d'autres possèdent un chromosome
linéaire. Il existe toutefois de rares bactéries possédant
deux chromosomes. La taille du génome s'exprime en millier de
nucléotides et peut être très variable selon les
espèces de bactéries.
L'analyse chimique de l'appareil nucléaire
indique qu'il est composé à 80 % d'ADN (le chromosome), à
10 % d'ARN et à 10 % de protéines [Carbonelle et al.,
2003].
· L'ADN (le chromosome) : Chez les bactéries
tout l'ADN est codant.
· L'ADN Extrachromosomique (les plasmides) : A
côté du chromosome, il peut exister des éléments
génétiques (ADN) de petites tailles (0,5 à 5 % du
chromosome bactérien), extra-chromosomiques, se sont les plasmides. Les
plus connus sont les plasmides de résistance aux antibiotiques, ils
portent des gènes
Introduction générale - 3 -
qui confèrent aux bactéries la
résistance à divers antibiotiques [Carbonelle et al.,
2003].
Dans le domaine des bactéries et en
particulier celui du Mycobacterium Tuberculosis, les séquences
complètes de génomes s'accumulent depuis 1995 (voir Tableau
0.1, Tableau 0.2, Tableau 0.3). Ces données ont
permis d'envisager l'étude du génome du Mycobacterium
Tuberculosis par des techniques informatiques, pour identifier et
connaître au mieux la source de l'infection afin d'aider les
spécialistes à trouver des solutions thérapeutiques et
stopper la diffusion de la bactérie et par conséquent stopper
l'épidémie par certains vaccins, ou antibiotiques.
Plusieurs approches informatiques notamment par la
fouille de données ont été alors développées
en exploitant des données biologiques en générale et de la
tuberculose en particulier, notamment par :
· l'utilisation d'algorithmes de recherche de
mots puis de couples de mots représentés énormément
dans les séquences ADN des souches et espèces
phylogénétiquement proches, ces séquences de lettres
particulières, permettent de repérer et d'identifier des
séquences anormales.
· la fouille de données génomiques
sans à priori pour faire émerger des sous-séquences d'ADN
qui peuvent donner des éléments d'informations sur les grandes
séquences d'ADN ;
· la recherche de gènes
co-régulés, etc.
En 1998, la première séquence
complète du génome de Mt H37RV a été
réalisée et a permis de dégager des
caractéristiques propres aux mycobactéries dont les plus
importantes [Carbonelle et al., 2003]:
· 51 % des gènes sont
dupliqués;
· 10 % du génome code pour 2 familles de
gènes qui codent eux mêmes pour 2 protéines nommées
PE et PPE;
· forte présence de séquences
répétées d'ADN, dont 65 copies de séquences
appelées MIRUs (Mycobacterial Interspaced Repetitive Unit), et de
répétitions directes appelées RDs. Ces séquences
répétées sont riches en particularités sur le
génome. Toutes ces caractéristiques de ce génome sont
autant chacune une source qu'on exploite en fouille de données
[Fleiishman et al., 2002], [Ferdinand et al., 2004], [Yokoyama et al.,
2007].
Introduction générale - 4 -
Problématique.
La représentation des séquences
biologiques. Dans un passé récent, la fouille de données
dans un contexte biologique utilisait la séquence dans sa structure
primaire à base de nucléotides (ex : AAGTCGTTGCTGGC) où
celle-ci est considérée comme une chaine de milliers de
caractères, en ce moment le gène, la protéine, et autres
éléments caractérisant n'étaient pas suffisamment
cernés (annotation incomplète) pour être exploités
efficacement et donc le prétraitement des données se basait
essentiellement sur des techniques de traitement de texte plus ou moins
aménagées selon le contexte.
Alors, nous avons envisagé un système
de fouille de données un peu plus élaboré du fait de
l'existence d'entités sémantiques dans le fichier de la
séquence en question (le
gène, la protéine, sa localisation, )
(voir Figure 0.2). Nous utilisons donc des
traitements
spécifiques pour obtenir une structure bien appropriée à
la fouille de données (voir Figure 0.3) ou les entités
sémantiques (gènes, protéines, ...) deviennent des
descripteurs, et on attribuera la valeur «0» en l'absence et
«1» en la présence de ce descripteur dans la
séquence.

Figure 0.2. Morceau de séquence
génomique Figure 0.3. Fichier des séquences
rapatriée de NCBI. ayant subi une
transformation.
Dans le cadre de cette étude, nous avons
développé des recherches sur les systèmes d'extraction de
règles d'association à partir des données (gènes,
...) [Chen et al., 2003], [Bahar et Chen, 2004], [Benabdeslem et al., 2007] et
nous avons réalisé un système baptisé BIODM :
BIOlogical Data Mining. En premier lieu, nous avons étudié
l'extraction de règles d'association en utilisant des algorithmes
appropriés. En deuxième lieu nous avons travaillé sur le
raffinement, des résultats, par un processus d'induction cellulaire BRI
(Boolean Rule Induction). Ce raffinement est assuré par une
modélisation booléenne.
Deux motivations concurrentes nous ont amenés
à adopter le principe des automates
Introduction générale - 5 -
cellulaires pour les systèmes à base de
règles d'association. En effet, nous avons non seulement souhaité
avoir une base de règles optimale (modélisation
booléenne), mais nous avons également exploité les
performances du moteur d'inférence cellulaire CIE de la machine
cellulaire CASI, déjà opérationnel [Benamina et Atmani,
2008].
Ce mémoire s'articule autour de quatre chapitres
:
Le chapitre I introduit l'extraction de la
connaissance à partir de données biologiques. Nous commencerons
par expliquer comment est né le besoin en fouille de données
biologiques et particulièrement en épidémiologie, ensuite
nous passerons en revue les différents types de données
biologiques auxquels nous seront amené à travailler pour donner
par la suite une vue d'ensemble du processus d'ECD biologiques que nous
envisageons de suivre. Une fois, toutes ces notions clarifiées nous
aborderons un état de l'art du domaine de l'ECD biologiques que nous
concluons par une étude comparative des méthodes et techniques
utilisées et une explication de notre contribution par cette
étude.
Le chapitre II aborde le principe de l'extraction des
règles d'association, une méthode descriptive de fouille de
données qui a reçu beaucoup d'intérêt en recherche.
Nous présentons le principe ainsi que les différents algorithmes
les plus en vue dans la littérature.
Le chapitre III est consacré à la
présentation du processus d'ECD biologiques que nous avons adopté
en particulier la modélisation booléenne des règles
d'association, résultat du module BRI, selon le principe de la machine
cellulaire CASI [Benamina et Atmani, 2008].
Le chapitre IV présente les données
expérimentales et l'architecture générale du
système que nous avons réalisé : BIODM. Ensuite, nous
présentons les résultats obtenus sur la base des
échantillons test que nous avons utilisés.
Finalement, nous concluons en synthétisant les
différentes étapes de notre contribution et nous proposons les
perspectives envisagées pour poursuivre cette recherche.
Chapitre I : L'extraction de connaissances à
partir de données biologiques - 6 -
Chapitre I.
L'extraction de connaissances à partir de
données biologiques
L'avènement des biotechnologies nouvelles a
permis, au cours des dernières années, d'améliorer les
connaissances sur le génome des agents pathogènes
épidémiologiques, de développer des moyens de lutte
efficace par le développement de plusieurs médicaments
appropriés. Par contre l'exploitation des données
génomiques n'a pas suivi le rythme des découvertes et
l'extraction de connaissances à partir de données (ECD)
biologiques, particulièrement à caractère
épidémiologique, s'est imposée d'elle-même afin de
répondre aux questions que se pose l'épidémiologiste comme
par exemple la recherche des facteurs de risque des maladies.
Ainsi et depuis le premier séquençage
d'une bactérie, des dizaines de génomes ont été
révélés. Les dispositifs expérimentaux tels que les
séquenceurs automatiques ont permis de constituer des banques de
données de séquences de génomes complets. Il fallait donc
analyser ces données, identifier les gènes, les protéines
produites et leurs fonctions pour comprendre les mécanismes cellulaires.
Les retombées de ces travaux sont énormes et concernent aussi
bien la biologie, l'épidémiologie et l'industrie pharmaceutique,
pour une meilleure compréhension des maladies et la découverte de
nouvelles réponses thérapeutiques.
I.1 Définition de l'extraction de connaissances
à partir de données biologiques
Le terme ECD (en anglais Knowledge Discovery in
Databases) est communément confondu avec la fouille de données ou
« Data Mining ». Ceci s'explique par le fait que la fouille de
données est l'étape principale du processus de l'ECD.
L'ECD a été définie comme suit
[Fayyad et al., 1996] : « l'ECD vise à transformer des
données (volumineuses, multiformes, stockées sous
différents formats sur des supports pouvant être
distribués) en connaissances. Ces connaissances peuvent s'exprimer sous
forme d'un concept général qui enrichit le champ
sémantique de l'usager par rapport à une question qui le
préoccupe. Elles peuvent prendre la forme d'un rapport ou d'un
graphique. Elles peuvent s'exprimer comme un modèle mathématique
ou logique pour la prise de décision. Les modèles explicites
quelle que soit leur forme, peuvent alimenter un système à base
de connaissances ou un système expert ».
Chapitre I : L'extraction de connaissances à
partir de données biologiques - 7 -
Cette définition apporte un concept nouveau,
celui de modèle et sous entend un autre celui de motif qui ne seraient
pas synonymes. En réalité il existe une différence entre
les deux :
Un modèle est une connaissance qui concerne la
totalité des données. Si le Data Miner possède un
modèle, il peut l'appliquer à chaque nouveau cas qui se
présente.
Un motif est une connaissance qui concerne une partie
des données. On ne peut l'appliquer à chaque nouveau cas. En
d'autres termes, c'est un modèle local, selon lequel se comporte une
partie des données et non pas la totalité.
I.2 Le processus de l'ECD biologiques
Avec le récent développement des
études à l'échelle génomique et protéomique,
les données biologiques se sont considérablement
multipliées et diversifiées. Ces données se
présentent alors sous la forme de séquences ou d'informations qui
proviennent de soumissions directes effectuées par les auteurs, par
l'intermédiaire d'Internet ou d'autres moyens électroniques
appropriés.
Nous trouvons alors des :
· des séquences et des données
d'expression de gènes (ADN, ARN, Protéines) ;
· des informations d'annotations (fonctions, ...)
de gènes et de protéines, etc.
Ces données biologiques sont stockées
dans des banques de données généralistes ou
spécialisées. On trouve alors des banques de données
:
· d'ADN : GenBank, DDBJ, EMBL, ...;
· d'ARN : RNAdatabases, QTL, ... ;
· de protéines : PIR ,Swiss-Prot, TrEMBL,
PDB, SCOP, ... ;
· de gènes : NCBI, dbEST, UniGene, Gis, ...
;
· ..etc.
L'ECD biologiques est un peu particulière
parce qu'en fait les données biologiques sont souvent dans un format
textuel (voir Figure 0.2) et ne se prêtent pas directement
à une exploitation par des systèmes classiques. Pour cela nous
présenterons ce processus dans son contexte biologique. Bien que le
processus général de l'ECD est particulièrement standard,
il présente néanmoins des traitements spécifiques d'une
étape à une autre et ce par rapport à la nature des
données traitées. Nous allons présenter une
démarche qui comprend les cinq étapes suivantes : la
sélection des données, le prétraitement, la
transformation, la fouille de données, l'évaluation et
l'interprétation des connaissances, en montrant d'une étape
à une autre, les particularités du processus d'ECD.
Chapitre I : L'extraction de connaissances à
partir de données biologiques - 8 -
(1) La sélection des
données
L'accès aux données se fait, dans notre
cas, à travers Internet via des interfaces spécialisées
pour le téléchargement d'échantillons expérimentaux
sélectionnés selon des critères fixés par
l'utilisateur. On utilise alors le système d'accès et de
récupération de données, ENTREZ de NCBI1.
Celui-ci permet d'interroger une collection de séquences disponibles
sous le format texte brut. Il permet aussi la recherche et l'extraction de
données relatives aux séquences nucléotidiques ou
protéiques, aux références bibliographiques
associées, et aux collections de séquences génomiques et
structurales, à l'aide d'une simple interrogation du serveur de NCBI
(National Center for Biotechnology Information).
Ensuite, ces données sont
récupérées sous la forme d'un ensemble de fichiers textes
bruts. À l'intérieur de ces fichiers, chaque séquence est
contenue dans une structure appelée « entrée »,
celle-ci comprend des informations liées à la séquence
considérée : structure, rôle biologique, organisme
d'origine...etc. Les données intéressantes sont stockées
au niveau de « champs » bien définis.
A l'intérieur de ces fichiers, la
donnée biologique peut être représentée sous
différents formats. Nous présentons les formats les plus
utilisés :
· FASTA (le format le plus simple)
· PIR (spécifique à la Bdd
PIR)
· STADEN
· Texte Brut.
Format FASTA
FASTA est sans doute le plus répandu et l'un
des plus pratiques. La séquence est décrite sous forme de lignes
de 80 caractères maximum, et précédée d'une ligne
de titre (nom, définition, ...) qui doit commencer par le
caractère ">". Plusieurs séquences peuvent être mises
dans un même fichier (voir Figure 1.1).
>entête de la séquence 1 Séquence
1
>entête de la séquence 2 Séquence
2
1
http://www.ncbi.nlm.nih.gov

Chapitre I : L'extraction de connaissances à partir de
données biologiques - 9 -
|
>gi|22777494|dbj|BAC13766.1| glutamate dehydrogenase
[Oceanobacillus iheyensis]
MVADKAADSSNVNQENMDVLNTTQTIIKSALDKLGYPEEVFELLKEPMRILTVRIPVRMDDGNV
LGGSHGRESATAKGVTIVLNEAAKKKGIDIKGARVVIQGFGNAGSFLAKFLHDAGAKVVAISDA
YGALYDPEGLDIDYLLDRRDSFGTVTKLFNNTISNDALFELDCDII
>EM|U03177|FL03177 FELINE LEUKEMIA VIRUS CLONE
FELV-69TTU3-16. AGATACAAGGAAGTTAGAGGCTAAAACAGGATATCTGTGGTTAAGCACCTG
GCCAGCAGTCTCCAGGCTCCCCA
|
Figure 1.1 : Exemple du format FASTA d'une
séquence protéique.
|
CODE
|
SIGNIFICATION
|
|
">"
|
Début de séquence.
|
|
gi|22777494
|
GenInfo Identifier
|
|
dbj|BAC13766.1|
|
Un enregistrement de séquence peut être
enregistré dans
plusieurs banques de données donc il y aura
un
identifiant dans la banque de données dans cet exemple c'est DNA
Database of Japan sous le n° dbj|BAC13766.1
|
|
BAC13766.1|
|
". 1" la séquence a été
révisée une fois
|
|
"glutamate dehydrogenase"
|
nom de la protéine
|
|
[Oceanobacillus iheyensis]
|
nom de l'organisme à partir duquel elle a
été déterminée.
|
Tableau 1.1 : Description du fichier FASTA de
l'exemple de la Figure 1.1.
Format STADEN

STADEN est le plus ancien et le plus simple. C'est une
suite de lettres par ligne terminée par un retour à la ligne (80
caractères maximum par ligne). Ce format n'autorise qu'une
séquence par fichier (voir Figure 1.2).
|
lovelace$ more zfmtsec
SESLRIIFAGTPDFAARHLDALLSSGHNVVGVFTQPDRPAGRGKKLMPSPVKVLAEEKGL
PVFQPVSLRPQENQQLVAELQADVMVVVAYGLILPKAVLEMPRLGCINVHGSLLPRWRGA
APIQRSLWAGDAETGVTIMQMDVGLDTGDMLYKLSCPITAEDTSGTLYDKLAELGPQGLI
TTLKQLADGTAKPEVQDETLVTYAEKLSKEEARIDWSLSAAQLERCIRAFNPWPMSWLEI
EGQPVKVWKASVIDTATNAAPGTILEANKQGIQVATGDGILNLLSLQPAGKKAMSAQDLL
NSRREWFVPGNRLV
|
Figure 1.2 : Exemple du format STADEN d'une
séquence protéique.
Format PIR
La première ligne commence par ">" suivi du
code de la séquence et du nom de la protéine. La deuxième
ligne contient une description textuelle de la séquence suivent
plusieurs lignes descriptives de la séquence elle-mêm,e et se
termine par une marque de fin de séquence "*" (voir Figure
1.3).

Chapitre I : L'extraction de connaissances
à partir de données biologiques -
10 -
|
>P1;1h7wa1
structureX:1h7wa1: 2 :A: 183 :A:undefined:undefined:
1.90:99.90
APVLSKDVADIESILALNPRTQSHAALHSTLAKKLDKKHWKRNPDKNCFHCEKLENNFD
DIKHTTLGERGALREACLKCADAPCQKSCPTHLDIKSFITSISNKNYYGAAKMIFSDNPLG
LTCGMVCPTSDLCVGGCNLYATEEGSINIGGLQQFASEVFKAMNIPQIRNPCLPSQEKMP*
|
Figure 1.3 : Exemple du format PIR d'une
séquence protéique.
|
CODE
|
SIGNIFICATION
|
|
|
|
">P1"
|
Début de la ligne
|
|
|
|
1h7wa1
|
Code de la protéine
|
|
|
|
structureX:1h7wa1: 2 :A: 183
:A:undefined:undefined: 1.90:99.90
|
description textuelle
séquence
|
de
|
la
|
|
"*".
|
Fin de la séquence
|
|
|
Tableau 1.2 : Description du fichier PIR de
l'exemple de la Figure 1.3.
Format Texte Brut
L'information biologique est décrite dans un
fichier au format texte brut ou chaque ligne a un sens bien précis,
comme par exemple, un code, un nom, etc. (voir Figure 1.4)

1: aac
aminoglycoside 2-N-acetyltransferase [Mycobacterium
tuberculosis CDC1551]
Other Aliases: MT0275
Annotation: NC_002755.2 (314424..314969,
complement)
GeneID: 923198
4270: tRNA-Pro-3
tRNA [Mycobacterium tuberculosis CDC1551] Annotation:
NC_002755.1 (4118796..4118872) GeneID: 922697
This record was discontinued.
Figure 1.4 : Exemple de fichier à
l'état brut de de la séquence génomique de
la souche MT
CDC1551 au format texte brut.
(2) Le prétraitement des
données
Le prétraitement consiste à nettoyer et
mettre en forme les données dans un formalisme approprié pour une
exploitation efficiente, i.e. l'élimination des données sans
importances particulières dans le processus d'ECD, et qui sont
susceptibles de réduire l'exactitude des modèles à
extraire. Ceci commence par un nettoyage des fichiers
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
11 -
par enlèvement des lignes inutiles, des termes
ou morceaux de texte, tels que n° ligne, caractères spéciaux
inutiles. La Figure 1.5 montre un morceau de séquence de
gène nettoyé, et la Figure 1.6, montre le
résultat final de cette étape.

1: aac
aminoglycoside 2-N-acetyltransferase [Mycobaterium
Tuberculosis CDC1551] GeneID: 923198
2: accD
acetyl-CoA carboxylase, carboxyl transferase, beta
subunit [Mycobaterium Tuberculosis CDC1551]
GeneID: 926242
Figure 1.5 : Morceau de la séquence
génomique nettoyée, de la souche Mt CDC1551.

aac |aminoglycoside 2-N-acetyltransferase | Mycobaterium
Tuberculosis CDC1551 | 923198
accD | acetyl-CoA carboxylase, carboxyl transferase,
beta subunit | Mycobaterium Tuberculosis CDC1551 | 926242
Figure 1.6 : Morceau de la séquence
génomique mise en forme, de la souche Mt CDC1551.
(3) La transformation des
données
Cette étape consiste à transformer les
données et les convertir en données appropriées (voir
Figure 1.6), pour exploitation. Ce sera une transformation vers un
formalisme base de données (attribut, valeur), à partir des
descripteurs possibles qui peuvent être dégagées à
cette étape. Ces descripteurs ou attributs vont aider à «
binariser » les entités dégagées et serviront ainsi
à alimenter une base de données.

aac |aminoglycoside 2-N-acetyltransferase | Mycobaterium
Tuberculosis CDC1551
| 923198
accD | acetyl-CoA carboxylase, carboxyl transferase, beta
subunit | Mycobaterium Tuberculosis CDC1551 | 926242
aceA-1 | isocitrate lyase |
Mycobaterium Tuberculosis CDC1551 | 923830
Figure 1.7 : Morceau de la séquence
génomique structurée, de la souche Mt CDC1551.
|
Séquence génomique
structurée
|
|
code_gene
|
nom_gene
|
id_gene
|
|
aac
|
aminoglycoside 2-N-
acetyltransferase
|
923198
|
|
accD
|
acetyl-CoA carboxylase, carboxyl transferase, beta
subunit
|
926242
|
|
aceA-1
|
isocitrate lyase
|
923830
|
Chapitre I : L'extraction de connaissances à
partir de données biologiques - 12 -
(4) La fouille de données
C'est le coeur du processus d'ECD biologiques.
L'extraction de connaissances est faite à partir de cette étape.
Elle consiste à dégager un ensemble de connaissances brutes
à partir des données prétraitées. Un exemple est la
recherche d'associations.
(5) Evaluation et interprétation des
connaissances
Dans la plupart du temps, les connaissances extraites
au terme de la précédente étape ne sont pas toutes
profitables. En effet, il est difficile d'avoir directement des connaissances
valides et utilisables par l'utilisateur humain, le Data Miner. Il existe, pour
la plupart des techniques de fouille de données, des méthodes
d'évaluation des modèles ou motifs extraits. Ces méthodes
peuvent aussi aider à corriger les modèles, et à les
ajuster aux données.
Selon le degré d'exactitude des connaissances
retournées par ces méthodes, l'expert du domaine décide
d'arrêter le processus d'ECD ou au contraire le reprendre à partir
d'une étape antérieure (le processus est itératif)
jusqu'à ce que les connaissances obtenues soient nouvelles,
interprétables, valides et utiles au Data Miner. Ce dernier peut les
utiliser directement ou les incorporer dans un système de gestion de
connaissances.
La Figure 1.8, ci-dessous montre l'aspect
itératif du processus, i.e. la possibilité de retourner à
n'importe quelle étape afin d'obtenir des connaissances de
qualité.
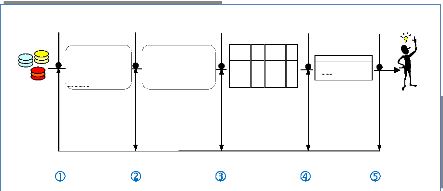
Banques de Données Biologiques (NCBI,
...)
Selection Prétraitement Transformation
Données biologiques cibles (séquence
génomique, séquence protéique,..)
1: aac
aminoglycoside 2-N-acetyltransferase [Mycobacterium tuberculosis
CDC1551]
Other Aliases: MT0275
Annotation: NC_002755.2 (314424..314969, complement) GeneID:
923198
Données Nettoyées et mises en
forme
accD | acetyl-CoA carboxylase, carboxyl transferase, beta
subunit | Mycobaterium Tuberculosis CDC1551 | 926242
aac |aminoglycoside 2-N-acetyltransferase | Mycobaterium
Tuberculosis CDC1551 | 923198
CDC1551 aac aminoglyco side 2-N-acetyltransf erase
Données Structurées
souche
Code gene
Nom gene
9231
98
Id gene
Fouille de données
aac -> ackA (75.0, 100.0) ackA -> aac (75.0,
100.0)
Motifs
Evaluation, interpretation
Connaissances
Figure 1.8 : Processus d'ECD
Biologiques.
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
13 -
I.3 Notre contribution
Dans notre étude, nous nous proposons
d'étudier les aspects physiologiques liés à la
génomique de cette bactérie modèle (Mycobacterium
Tuberculosis), à savoir : les gènes, protéines, et autres
données génomiques, ceci afin de mieux connaitre notre terrain
expérimental.
Ensuite, nous étudions les outils de l'ECD pour
les utiliser sur les données précitées, et dégager
une approche expérimentale par des moyens informatiques afin de donner
à l'expert du domaine des connaissances qui permettent d'avoir quelques
éléments de réponses possibles aidant à comprendre
certains processus biologiques et se fixer de nouvelles hypothèses et
continuer ainsi le processus de compréhension et de recherche
médicale.
En premier
Nous avons établi un état de l'art de
l'ECD, où nous présentons les techniques de l'ECD avec certains
détails d'une technique à une autre et qui ne sont pas forcement
en rapport direct avec notre étude. Ceci est justifié par le fait
que ce travail est notre première contribution dans ce domaine, nous
avons alors pensé qu'il est utile de faire le tour du domaine afin de
situer la technique de recherche d'associations parmi les différents
outils de l'ECD. Ensuite, une étude comparative des différentes
méthodes existantes a été faite pour nous positionner par
rapport à celles-ci et nous fixer sur la plus appropriée pour
notre étude.
Deuxièmement
Nous avons abordé l'étude de l'agent
pathogène afin de cerner la nature et le type de données
biologiques qui nous intéressent et ainsi pouvoir localiser d'où
puiser nos données expérimentales pour la fouille de
données, i.e. les sources de données biologiques relatives au
Mycobacterium Tuberculosis.
Troisièmement
Nous avons établi notre propre démarche
expérimentale par un processus d'ECD pour générer des
connaissances à partir de données biologiques. Ces connaissances
vont êtres profitables et exploitables à deux niveaux
:
· profitables au premier niveau au
spécialiste du domaine pour la compréhension des aspects
biologiques liés à la pathologie ;
· exploitables au deuxième niveau par la
machine cellulaire pour l'inférence et la
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
14 -
déduction.
Le processus informatique établi procède
en deux étapes : une fouille de données est faite dans un premier
temps et donnera des règles d'association, ensuite ces règles
sont transformées dans un deuxième temps pour produire des
règles booléennes inductives qui vont alimenter la base de
connaissances de la machine cellulaire CAST, [Atmani et Beldjilali, 2007],
[Abdelouhab et Atmani., 2008], [Benamina et Atmani, 2008]. Cette machine a
été développée pour l'acquisition automatique
incrémentale de connaissances par induction et la prédiction par
déduction [Atmani et Beldjilali, 2007].
Ainsi, notre contribution a adopté la
démarche suivante :
(1) Etude et sélection des données
biologiques relatives au mycobactérium tuberculosis ;
(2) Extraction des motifs fréquents et
recherche des règles d'association ;
(3) Production des règles booléennes
inductives pour la machine cellulaire CAST.
I.4 Etat de l'art de l'ECD biologiques
Vu la variété des données
biologiques et par la même des banques de données biologiques,
différents travaux de fouilles de données biologiques ont
été faits. Nous présentons quelques uns mais la liste
n'est pas exhaustive, et nous mentionnons quand même que ce qui a
été fait à nos jours l'a été à base
de séquences biologiques ou de cohortes et suivent deux orientations
principales : certains travaux touchent directement
l'épidémiologie alors que d'autres la touchent indirectement
(génomique et protéomique), mais sont d'un grand apport pour la
compréhension des maladies et par la même des
phénomènes épidémiologiques. Nous présentons
les travaux réalisés dans un tableau récapitulatif (voir
Tableau 1.4) qui donne une vue générale sur les
principales méthodes de fouille de données utilisées en
biologie et dans le domaine des maladies. Ce tableau donne une tendance
à priori sur les méthodes de fouille utilisées mais notons
quand même que c'est un tableau qui résume des articles que nous
avons pu lire et se rapportant à notre sujet sans pour autant
prétendre à l'exhaustivité, vu les travaux qui se font
continuellement.
La classification de protéines
Le prétraitement des séquences biologiques
permet de préparer les données et de les
I.4.1 La fouille de séquences biologiques
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
15 -
exploiter par la suite afin d'identifier des
gènes, comparer des séquences, rechercher des motifs, ou
détecter des signaux (segment ADN) qui indiquent à la cellule la
protéine qui doit être exportée ou
sécrétée. Pour la classification des protéines on
commence souvent par cette phase de prétraitement comme indiquée
pour construire des attributs plus significatifs que la chaîne de
caractères originale. Différentes approches ont été
étudiées allant de l'extraction des descripteurs (variables qui
décrivent au mieux) discriminants, à l'extraction des n-grammes
[Mhamdi et al., 2006], à d'autres en utilisant les modèles de
Markov cachées [Hergalant et al., 2005].
La prédiction de
gène
Les modèles de Markov cachés
interviennent dans de nombreux algorithmes d'analyse de séquence, que ce
soit pour la détection de gènes, l'inférence et la
détection de motifs, ou encore la recherche de mots exceptionnels.
L'utilisation des modèles de Markov cachées d'ordre 2 (HMM2) est
d'une grande utilité dans la segmentation de grandes séquences
ADN en grandes classes déduites à partir de courtes
séquences riches en sémantiques biologiques. Cette segmentation
est utilisée pour prédire des ensembles de gènes
co-régulés donc susceptibles d'avoir des fonctions liées
[Hergalant et al., 2002].
La segmentation de séquences
biologiques
Les très grandes séquences d'ADN qui
constituent les génomes ne sont pas avantageuses directement pour la
fouille de données, donc il est nécessaire de dégager des
sous séquences d'ADN significatives pour une exploitation en
génomique fonctionnelle. Les modèles de Markov cachés ont
étés utilisés pour permettre la segmentation de grandes
séquences d'ADN en différentes classes pour l'étude d'une
bactérie du sol du genre Streptomyces, important producteur
d'antibiotiques. En effet, l'ADN est stratifiée en différents
niveaux de structures, de la plus globale qui reflète une organisation
propre à une espèce considérée, aux portions
locales qui décrivent des constituants fonctionnels particuliers, en
passant par des régions intermédiaires qui délimitent des
domaines où réside une certaine homogénéité
(par exemple pour les protéines).
Recherche de similitudes et étude
d'alignement
La recherche de similarité de séquences
ou de structures est un autre sujet abordé avec les méthodes de
comparaison des séquences ADN et de protéines. Cette comparaison
peut être en local c'est-à-dire entre séquences (Alignement
Local) ou globale (Alignement Global) avec une base de données. Elle a
pour but le repérage des
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
16 -
endroits où se trouvent des régions
identiques ou très proches entre deux séquences et déduire
celles qui sont significatives et qui correspondent à un sens
biologique. En général les algorithmes fonctionnent sur des
segments de séquences sur lesquels on regarde s'il existe ou pas une
similitude significative et la comparaison de séquences s'appuie sur une
de ces trois notions : la recherche de segments identiques, de segments
similaires, ou d'alignements. Leur but est de filtrer les données de la
banque en étapes successives, car peu de séquences vont avoir des
similitudes avec la séquence comparée. Ces programmes calculent
ensuite un score pour mettre en évidence les meilleures similitudes
locales qu'ils ont observées. Plusieurs programmes ont
étés créés dont les plus connus et les plus
utilisés par les biologistes sont les logiciels FASTA et BLAST [Chervitz
et al., 1999].
La recherche de motifs
fréquents
La recherche de séquences
répétées (motifs fréquents), en tandem ou
répétées en dispersion sur l'ensemble du génome,
utilise les Modèles de Markov cachées d'ordre 2. Une étude
a été faite en utilisant cette méthode pour classer un
résidu ou un groupe de résidus nucléotidiques [Hergalant
et al., 2002].
La recherche
d'hétérogénéité
L'analyse de séquences génomiques
à l'aide des modèles de Markov cachés a montré une
grande capacité à détecter des zones
hétérogènes dans les génomes, ces zones qui peuvent
renseigner sur l'implication d'un pathogène donné dans une
maladie [Hergalant et al., 2002].
La recherche de séquences
exogènes
Un autre aspect, de l'étude des gènes,
est le transfert horizontal (échange de matériel
génétique entre bactéries). Des études ont
été faites sur le genre Streptococcus parce qu'il revêt une
importance particulière en renfermant les bactéries
pathogènes. L'utilisation des modèles de Markov cachés
[Hergalant et al., 2002], a permis d'identifier certaines séquences
exogènes au sein de ces génomes qui sont susceptibles de contenir
des gènes de virulence, caractéristiques d'un pathogène,
ou des gènes d'adaptation écologique particulière, ces
résultats montrent que des bactéries seraient en mesure de
transmettre des gènes de virulence à d'autres bactéries
encore inoffensives. Ce genre de découverte améliore la
compréhension du phénomène de résistance à
un antibiotique.
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
17 - I.4.2 fouille du génome
De nombreuses maladies, rares ou fréquentes,
dont souffrent les humains ont une origine génétique tels que le
diabète, les maladies cardiovasculaires ou la mucoviscidose [Maumus et
al., 2005]. Ces maladies ont un caractère plus ou moins
héréditaire dans le sens où des anomalies chromosomiques
sont à l'origine ou favorisent l'apparition de ces maladies. Cela
signifie qu'un ou plusieurs gènes anormaux sont là où est
la maladie. L'étude du génome ou la génomique structurelle
(recherche de mutation, de délétion, ...) peut montrer des
anomalies tout comme la génomique fonctionnelle (compréhension du
fonctionnement des gènes et des autres composantes du génome).
Cette étude se situe à plusieurs niveaux depuis l'échelle
nucléique jusqu'à celui de la génomique comparative avec
pour objectif la localisation, de structures, de régions fonctionnelles,
la caractérisation d'une fonction biologique ou la prédiction
d'autres gènes, c'est ce qui explique le décryptage du
génome par plusieurs travaux de fouille dont les suivants :
Identification de gènes codants et non
codants
Si l'existence de facteurs génétiques de
susceptibilité est fortement soupçonnée dans de nombreuses
maladies, leurs identifications sont souvent difficiles ce qui ouvre la voie
à une étude de recherche des facteurs génétiques de
susceptibilité aux maladies à l'échelle de tout le
génome. Sur le volet de l'annotation des génomes,
c'est-à-dire l'identification des gènes codants (exon), et non
codants (intron), la localisation du début des gènes ainsi que la
détection de petits gènes restent difficiles, et dans le but de
répondre à ces problèmes, on utilise des modèles de
Markov cachés couplés à une estimation par maximisation de
la vraisemblance (comptage des mots de m nucléotides) [Prum et al.,
2001].
La classification et l'identification de
gènes candidats
L'analyse de séquences sur puces à ADN
permet d'obtenir des morceaux de séquences appelées EST
(Expressed Sequence Tags) courtes, de 300-500 nucléotides en
général, ces éléments permettent d'identifier les
variations génétiques associées (zone de
susceptibilité) comme par exemple le gène NOD-2 impliqué
dans la maladie de CROHN.
L'approche gène candidat recherche et met en
relation des gènes nouveaux, découverts par le
séquençage, avec les pathologies orphelines (dont on ne
connaît pas encore le gène responsable) ou des pathologies
complexes, (obésité, arthrose, ...). Deux
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
18 -
classifications ont été
utilisées, l'une pour classer les patients dont la maladie se ressemble
quant à l'expression des gènes, et l'autre qui cherche les
gènes ou les ensembles de gènes dont l'expression est
différente chez des patients de mauvais et de bon pronostic [Maumus et
al., 2005].
I.4.3 Fouille de cohortes
Les cohortes fournissent aussi un tas de
données médicales et en particulier biologiques, qui permettent
de travailler directement sur des cas réels (sujets exposés, non
exposés) et permettent de montrer le rôle et la contribution des
facteurs génétiques et environnementaux dans une maladie. Les
données recueillies sont de 3 types : cliniques, biologiques, et
génétiques.
Les données cliniques. Se divisent en examens
cliniques systématiques (taille, poids,
pression artérielle, ....), et en examens
cliniques spécifiques (échographie, ).
Les données biologiques. Sont les dosages
systématiques réalisés, la biochimie, NFS «
numération de formule sanguine » et analyse d'urine.
Les données génétiques. Se
rapportent à chaque sujet.
On dit que le sujet est génotypé, si
l'on procède par le recueillement de toutes ces données
génétiques. Ce génotypage permettra d'avoir des SNPs
(Single Nucleotide Polymorphisms ou polymorphismes génétiques)
correspondant à tous les processus métaboliques impliqués
dans la maladie. Une fois ces données recueillis, les premières
expérimentations d'extraction de règles sont utilisées
pour permettre la détection d'interactions de type
gène-gène et gène-environnement. Les méthodes de
classification sont aussi très utilisées pour comprendre les
maladies, une étude comparative des différents algorithmes a
été faite pour la pertinence de ces méthodes [Chervitz et
al., 1999], et a montré que les réseaux de neurone, et les SVM
donnent à peu près les mêmes résultats
comparativement aux K-ppv et les arbres de décision. Ces méthodes
de classification sont aussi utilisées pour étudier la
variété des segments ADN et leurs relations avec des maladies ou
symptômes particuliers [Etienne, 2004].
L'extraction de motifs fréquents et
recherche d'association
La technique d'extraction de motifs fréquents
pour l'extraction de règles d'association et de profils
génétiques pour une maladie a bien montré de bons
résultats. Une étude dans ce sens a été faite sur
la cohorte STANISLAS [Maumus et al., 2005], pour la compréhension de
l'athérosclérose et étudier les mécanismes
physiopathologies du syndrome métabolique et déterminer les
facteurs influents. Cette technique a permis
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
19 -
aussi de mettre en évidence la relation entre
un gène en l'occurrence le HLA-DQ impliqué dans les maladies
auto-immunes tel que la maladie coeliaque sur une cohorte de 470 individus
(témoins) de 3 pays européens [Maumus et al., 2005]. Aussi,
l'apprentissage des règles à partir de données d'une
étude épidémiologique « cas-témoins » a
montré une grande utilité pour la compréhension de
certaines maladies telle que le cancer du nasopharynx de 1289 observations.
L'objectif était de connaître les différents facteurs
impliqués dans cette maladie [Benabdeslem et al., 2007].
L'association entre un facteur d'exposition et la
maladie en question a été aussi utilisé pour trouver les
facteurs liés à une maladie, identifier un facteur d'exposition,
mettre en évidence le lien entre la maladie et ce facteur avec un test
statistique approprié, et ensuite mesurer la force de ce lien avec des
indicateurs bien connus en épidémiologie tels que le RR (Risque
relatif), DR (Différence de Risque) et OR (Odds Ratio) [Boutin et al.,
2003].
La recherche de gènes
candidats
Pour les maladies multifactorielles (facteurs
environnementaux et génétiques) la part du
génétique met souvent en jeu plusieurs gènes de
susceptibilité [Etienne, 2004], [Bahar et Chen, 2004], ce sont ces
gènes qui sont mis en évidence par une approche statistique
exploratoire utilisant les modèles de Markov cachés sur une
cohorte. En effet la comparaison des génomes, dans le cadre d'une
étude de cohorte familiale consiste à vérifier le partage
génétique le long de chaque chromosome. La description au travers
d'un modèle probabiliste markovien de ce partage génétique
permet de déterminer les gènes impliqués dans une maladie
[Maumus et al., 2005].
La recherche de gènes candidats et de
marqueurs génétiques
L'épidémiologie génétique
a tenté de comprendre le déterminisme génétique
(approche gène candidat / maladie) par l'approche des marqueurs
génétiques dans des familles de malades tels que le cancer par
exemple. Cette approche a pu montrer d'une part l'existence de facteurs
génétiques et les identifier. La stratégie était de
tester le rôle éventuel de gènes candidats en utilisant des
marqueurs situés sur ces gènes. Un gène candidat est un
gène dont la fonction est soupçonnée dans le processus
étiopathogénique. Il a été nécessaire de
présenter les liens (associations) existants entre les gènes et
les maladies génétiques. Une constatation à
été faite : les quelques trois mille maladies
génétiques connues peuvent recouvrir deux cas : les maladies mono
géniques (dues à un seul gène) et les maladies
polygéniques et/ou polyfactorielles.
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
20 -
Prédiction de maladies
L'utilisation des réseaux bayésiens a
été très utile pour la prédiction des risques de la
maladie coronarienne, une étude a été faite dans ce sens
sur une population de 8000 participants, et a montré de bons
résultats de prédiction de cette maladie [Maumus et al.,
2005]
I.5 Les méthodes de fouille de
données
La fouille de données n'est ni un outil, ni une
technique, c'est plutôt une science encapsulant des méthodes dont
le but est d'extraire des connaissances à partir de données assez
structurées. Chaque méthode vise un objectif, i.e. une
tâche, qui n'est réalisable par une seule méthode, mais
plus tôt par un ensemble de méthodes complémentaires,
chacune choisie en fonction du domaine d'application, du type des
données à analyser, et du type des connaissances que nous voulons
extraire.
Une tâche correspond donc à l'objectif du
Data Miner, ainsi par exemple la classification d'objets, à partir de
plusieurs caractéristiques connues, est une tâche de fouille de
données. Les tâches peuvent être catégorisées
de plusieurs façons. D'ailleurs, il n'existe pas de taxonomie standard
dans la littérature. Fayyad et al divisent les tâches en deux
[Fayyad et al., 1996] : les tâches descriptives et les tâches
prédictives. D'autres, les divisent en méthodes symboliques et
numériques, supervisées ou non supervisées et par
renforcement. D'autres auteurs encore, divisent les tâches de fouille de
données en sept types [Goebel et Gruenland, 1999]: la prédiction,
la régression, la classification, le groupement, l'analyse de liens, les
modèles de visualisation et l'analyse exploratoire de
données.
Comme Il n'existe pas de taxonomie standard nous avons
donc choisi de décrire les trois approches qui reviennent souvent dans
la littérature et de présenter ensuite une catégorisation
la plus adaptée.
1ere approche (description,
prédiction)
Une distinction est faite selon l'objectif. On a alors
les méthodes descriptives (caractérisation, discrimination,
extraction de règles d'association, analyse de cas extrême) et les
méthodes prédictives (classification, groupement).
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
21 - 2eme approche (symbolique,
numérique)
Une distinction est faite par type de données
traitées en entrée. Nous distinguons alors :
· les méthodes symboliques, notamment
l'extraction de motifs fréquents, l'extraction de règles
d'association, la classification par treillis, et la classification par arbres
de décision ;
· les méthodes numériques, en
particulier, les méthodes statistiques (classification automatique,
ACP), les modèles de Markov cachés d'ordre 1 et 2, les
méthodes neuronales, et les algorithmes
génétiques.
3eme approche (supervisé, non
supervisé, par renforcement)
L'apprentissage supervisé est basé sur
la construction d'un modèle où l'on considère un ensemble
de classes d'exemples de cas déjà traités, et on cherche
l'appartenance d'un objet à une classe en considérant les
similarités entre l'objet et les éléments de la classe.
Ces classes sont nommées préalablement. On essayera d'expliquer
et/ou de prévoir un ou plusieurs phénomènes observables et
effectivement mesurés.
L'apprentissage non supervisé utilise un
ensemble de données dans lequel aucune d'elles n'a d'importance
particulière par rapport aux autres, i.e. aucune n'est
considérée individuellement comme l'objectif de l'analyse. Cette
méthode permet la formation de groupes d'éléments
(clusters) homogènes partageant certaines similarités. Il n'y a
pas de classes à expliquer ou de valeur à prédire, la
méthode doit découvrir par elle-même les groupes
d'éléments. Il appartiendra ensuite à l'expert du domaine
de déterminer l'intérêt et la signification des groupes
constitués.
L'apprentissage par renforcement a pour but
d'apprendre à partir d'expériences acquises en différentes
situations, de façon à optimiser une récompense
numérique au cours du temps. Un exemple est de considérer un
agent autonome qui doit prendre des décisions en fonction de son
état courant. En retour, l'environnement récompense l'agent en
positif ou négatif. L'agent cherche un comportement décisionnel
(stratégie ou politique) optimal pour maximiser la somme des
récompenses au cours du temps.
Pour notre étude, nous avons donc choisi la
catégorisation citée dans l'article de Larose [Larose, 2005] et
nous avons fait une étude comparative sur cette base, car nous pensons
que relativement à celles qui existent dans la littérature c'est
la plus utilisée. Nous avons alors les tâches suivantes
:
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
22 -
(1) La description
La description explore les données sans
forcément chercher à prouver une hypothèse ou
répondre à une question. L'objectif est de décrire les
données ou donner des résumés sur celles-ci. On utilise
pour cela, les outils d'analyse exploratoire de données (EAD) qui sont
interactifs et visuels et s'appuient sur des méthodes graphiques. Viens
ensuite, le Data Miner qui interprétera les modèles
déduits.
(2) L'estimation
Elle consiste à estimer la valeur d'un champ
à partir des caractéristiques d'un objet. Le champ à
estimer est un champ à valeurs continues. Contrairement à la
classification, le résultat d'une estimation permet d'obtenir une valeur
pour une variable continue. Par exemple, on peut estimer le revenu d'un
ménage selon divers critères (type de véhicule,
profession, type d'habitation, etc.), il sera ensuite possible de
définir des tranches de revenus pour classifier les individus. Un
intérêt de l'estimation est de pouvoir ordonner les
résultats pour ne retenir, si on le désire, que les n meilleures
valeurs.
(3) La prédiction
Elle consiste à estimer la valeur future d'un
champ à partir des caractéristiques d'un objet. Cela revient
à prédire la valeur d'une variable à partir d'autres
variables, dont on connaît les valeurs, appelées variables
prédictrices. Dans ce cas, la construction des modèles se fait
grâce à l'apprentissage à partir d'anciens cas
(données dont on connaît les valeurs de la variable à
prédire). Chaque modèle construit est accompagné par une
valeur d'une fonction score indiquant son taux d'erreur (ou son taux
d'exactitude).
(4) La classification
Elle consiste à examiner les
caractéristiques d'un élément qui se présente afin
de l'affecter à une classe prédéfinie ou connue à
l'avance, celle-ci est un champ particulier à valeurs discrètes.
L'utilisation de la classification se fait depuis déjà bien
longtemps pour comprendre notre vision du monde (espèces animales,
minérales ou végétales). Elle permet de repartir en
classes des objets ou individus. Ces classes sont discrètes :
homme/femme/bleu/blanc, etc. Un exemple de classification est l'attribution
d'un sujet principal à un article de presse.
Dans un cadre informatique, les objets ou
éléments sont représentés par un enregistrement et
le résultat de la classification viendra alimenter un champ. Il est
souvent utilisé les méthodes suivantes : la méthode K-ppv
(plus proche voisin), les arbres de décision, et les réseaux de
neurones.
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
23 -
(5) Le groupement
Pour cette tâche, il n'y a pas de classe
à expliquer ou de valeur à prédire définie à
priori, il s'agit de créer des groupes d'objets homogènes dans la
population, ayant les mêmes caractéristiques.
(6) La recherche d'association
Elle consiste à trouver des corrélations
ou relations entre attributs (items). La recherche de liens associatifs est
utilisée intensivement dans les bases transactionnelles. Il s'agit de
découvrir les items les plus fréquemment en relation. Un exemple,
est la détermination des articles (cahier et stylo bleu, riz et oeufs,
...) qui se vendent ensemble dans un supermarché.
Ainsi, pour réaliser l'une ou l'autre des
tâches précitées, on utilise une ou plusieurs
méthodes et elles sont nombreuses. Il est difficile de les
présenter toutes. Ceci dit, celles qui sont présentées
ci-dessous sont tout de même les plus importantes. Il faut noter qu'il
n'y a pas de méthode meilleure, chacune présente des avantages et
des inconvénients.
(1) Les méthodes de
visualisation
Elles permettent la visualisation des données
et donc l'analyse l'exploratoire avec comme objectif le dégagement de
motifs, de structures, de synthèses, etc. Elles sont basées sur
des graphiques qui facilitent l'interprétation des résultats.
Dans le premier type de visualisation, l'utilisateur ne connaît pas
forcément ce qu'il cherche dans les données, il essaye de
chercher des modèles, des motifs, ou plus généralement des
hypothèses qu'il veut démontrer. Au deuxième type de
visualisation, l'utilisateur a une hypothèse qu'il veut tester et
confirmer. Ce type de visualisation est dérivé directement des
statistiques, et ne convient pas vraiment au principe même de la fouille
de données (bien qu'il en fasse partie). Les méthodes les plus
utilisées sont : les graphiques de statistiques
élémentaires (moyenne, écart type, variance), les
histogrammes, les nuages de points, et les courbes.
(2) Les réseaux de neurones
Un réseau neuronal est composé de
groupes de noeuds (neurones) où chaque groupe de noeuds correspond
à une couche. Il est formé par au moins trois couches : input,
intermédiaire et output. Dans la couche input, chaque noeud correspond
à une variable prédictrice. La couche output contient un ou
plusieurs noeuds et les variables à prédire.
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
24 -
Le réseau peut avoir plusieurs couches
intermédiaires (mais un seul input et un seul output), appelées
aussi couches cachées. Chaque noeud de la couche j est relié
à tous les noeuds de la couche j+1. A chaque arc est associé un
poids (une valeur) Wij, c'est le poids de l'arc entre le noeud i et le noeud j.
Les valeurs input des noeuds de la première couche sont les valeurs des
variables prédictrices. Les valeurs internes des autres noeuds (des
couches intermédiaires et de la couche output) sont calculées
à travers une fonction de sommation.
Les réseaux de neurones sont des outils
très utilisés pour la classification, l'estimation, la
prédiction et le groupement. Ils permettent de construire un
modèle qui prédit la valeur d'une variable à partir
d'autres variables connues appelées variables prédictrices. Si la
variable à prédire est discrète (qualitative) alors il
s'agit d'une classification, si elle est continue (quantitative) il s'agit
alors de régression. Les méthodes les plus utilisés sont :
le Perceptron multicouches et les réseaux de Kohonen.
(3) Les arbres de décision
Un arbre de décision est une
représentation graphique d'une procédure de classification. Les
noeuds internes de l'arbre sont des tests sur les champs ou attributs, et les
feuilles sont les classes. Lorsque les tests sont binaires, le fils gauche
correspond à une réponse positive au test et le fils droit
à une réponse négative.
Un arbre de décision cible la classification
i.e. la prédiction de variables discrètes. La méthode
consiste à construire un arbre et pour chaque élément
qu'on veut classifier, il entre par le noeud racine et passe d'un noeud
père à un noeud fils, s'il satisfait une condition posée.
Le noeud feuille auquel il arrivera est sa classe. Un arbre de décision
peut donc être perçu comme étant un ensemble de
règles qui mènent à une classe. La construction de l'arbre
se fait par un algorithme approprié. Les algorithmes les plus
utilisés sont : ID3, CHAID, CART, QUEST, et C5 [Quinlan, 1986],
[Wilkinson, 1992], [Gaussier, 2009], [Quinlan, 1993], [Denison et al., 1998],
[Callas, 2009].
(4) SVM (machines à vecteur de
support)
Les machines à vecteurs de support sont une
classe d'algorithmes d'apprentissage initialement définis pour la
discrimination i.e. la prévision d'une variable qualitative initialement
binaire. Elles ont été ensuite généralisées
à la prévision d'une variable quantitative. Dans le cas de la
discrimination d'une variable dichotomique, ils sont basés sur la
recherche de l'hyperplan de marge optimale qui, lorsque c'est possible, classe
correctement les données tout en étant le plus
éloigné possible de toutes les
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
25 -
observations. Le principe est donc de trouver une
fonction de discrimination, dont la capacité de
généralisation (qualité de prévision) est la plus
grande possible. Donc les SVM peuvent être utilisés pour
résoudre des problèmes de discrimination (i.e. décider
à quelle classe appartient un échantillon), ou de
régression (i.e. prédire la valeur numérique d'une
variable).
(5) K - plus proches voisins
(K-ppv)
La méthode des K plus proches voisins (K-ppv,
nearest neighbor ou K-nn) est une méthode dédiée à
la classification qui peut être étendue à des tâches
d'estimations. La méthode K-ppv est une méthode de raisonnement
à partir de cas. Elle part de l'idée de prendre des
décisions en recherchant un ou des cas similaires déjà
résolus en mémoire. Elle décide de la classe à
laquelle appartient un nouveau cas, en examinant les k cas qui lui sont les
plus similaires ou proches. Il n'y a pas d'étape d'apprentissage
consistant en la construction d'un modèle à partir d'un
échantillon d'apprentissage. C'est l'échantillon d'apprentissage
qui constitue le modèle. On lui associe une fonction de distance et une
fonction de choix de la classe en fonction des classes des voisins les plus
proches.
(6) Les réseaux
bayésiens
Un réseau bayésien ou probabilistique
est un graphe dirigé, acyclique où chaque noeud représente
une variable continue ou discrète, et chaque arc représente une
dépendance probabilistique. Si un arc relie un noeud Y à un noeud
Z, alors Y est le parent de Z et Z est le descendant de Y. Chaque variable est
indépendante des variables auxquelles elle n'est pas reliée. Les
variables peuvent être continues ou discrètes. Chaque lien entre
deux variables est pondéré par la valeur de la dépendance
probabilistique, ainsi la valeur que porte l'arc reliant Y à Z est en
fait P (Z/Y).
(7) Induction de règles
C'est une technique qui permet d'identifier des
profils, associations ou structures entre les items ou objets qui sont
fréquents dans les bases de données. Autrement dit, il s'agit
d'identifier les items qui apparaissent souvent ensemble lors d'un
évènement. Cette règle d'association est une règle
de la forme : « Si X et Y alors Z », règle dont la
sémantique peu être énoncée : « Si X et Y
apparaissent simultanément alors Z apparait ». Pour
considérer et exprimer cette association sous forme d'une règle,
il faut définir des quantités numériques qui vont servir
à valider son intérêt, d'où : le support et la
confiance.
Le support est la fréquence d'apparition
simultanée des éléments qui apparaissent
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
26 -
dans la condition et dans le résultat, soit :
support=fréquence (condition et résultat), et la
confiance=fréquence (condition et résultat) / fréquence
(condition). Ainsi, les règles dont le support et la confiance sont
assez élevés sont alors privilégiées. Les
algorithmes les plus utilisés sont : Apriori, AIS, FP-Growth [Agrawal et
al., 1993],[Agrawal et Srikant., 1994], [Han et al., 2000].
(8) Les techniques de groupement
Ces techniques ou méthodes visent la
description des données. Elles consistent à diviser les
données en groupes de manière à ce que les données
d'un même groupe soient similaires et les données de deux groupes
différents soient différentes. Le degré de ressemblance
entre les données est calculé par une méthode de
similitude. Les méthodes de groupement se divisent en deux types : le
groupement basé sur les partitions et le groupement hiérarchique.
La technique la plus connue du premier type est la méthode des
K-moyenne. Elle consiste à diviser les données en K groupes, K
étant donné par l'utilisateur. Cette méthode commence par
un groupement aléatoire des données (en K groupes), ensuite
chaque enregistrement est affecté au plus proche groupe. Après
l'exécution de la première itération, les moyennes des
groupes sont calculées et le processus est répété
jusqu'à stabilisation des groupes. L'algorithme le plus utilisé
est : K-moyenne.
(9) Les modèles de Markov
cachés
Les modèles de Markov cachés d'ordre 1
ou 2 (HMM1 et HMM2 pour Hidden Markov Models) sont utilisés pour la
classification des différentes données temporelles ou spatiales.
Contrairement aux algorithmes classiques qui fournissent une réponse
exacte, les HMMs permettent un apprentissage automatique, ils interviennent par
exemple dans de nombreux algorithmes d'analyse de séquences biologiques
que ce soit pour, la détection de gènes, l'inférence et la
détection de motifs, ou encore la recherche de mots exceptionnels. En
effet, pour toutes ces opérations sur les séquences biologiques,
il est tenu compte des caractéristiques propres de la séquence,
de la position d'une sous séquence i.e. des caractéristiques de
ses voisins temporels ou spatiaux.
(10) Les treillis
Les treillis de concepts formels (ou treillis de
Galois) sont une structure mathématique permettant de représenter
les classes non disjointes sous-jacentes à un ensemble d'objets
(exemples, instances, t-uples ou observations) décrits à partir
d'un
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
27 -
ensemble d'attributs. Ces classes non disjointes sont
aussi appelées concepts formels. Une classe matérialise un
concept. Ce concept peut être défini formellement par une
extension (exemples du concept) ou par une intension (abstraction du concept).
Les treillis sont utilisés en classification.
(11) Autres méthodes
La régression linéaire
(méthode statistique)
C'est une technique qui vise la prédiction de
la valeur d'une variable continue. Son objectif est de définir le
meilleur modèle qui associe une variable quantitative « output
» à plusieurs variables prédictrices « input ».
Cette opération s'appelle ajustement du modèle aux
données. Les modèles linéaires sont les plus
fréquemment utilisés. C'est ce qu'on appelle la régression
linéaire. La relation qui relie une variable à prédire
Y à p autres variables prédictrices est une
équation de régression souvent sous cette forme : Y = a0 + a1
X1 + a2 X2 + ... + ap Xp
Les méthodes les plus utilisés sont :
régression simple, régression multiple.
Les algorithmes
génétiques
Les algorithmes génétiques ne
constituent pas une méthode de fouille de données à part
entière et ne ciblent directement aucune tâche. Ils viennent aider
le processus de fouille de données. Ce sont des heuristiques qui guident
la recherche de bons modèles dans un espace de recherche très
vaste. Les algorithmes génétiques se basent sur les principes de
sélection, enjambement, et mutation qui sont des notions issues de la
génétique.
I.6 Etude comparative
Après avoir passé en revu les
différentes tâches et méthodes, nous avons établi un
tableau récapitulatif (voir Tableau 1.4), mentionnant les
principales caractéristiques des différentes méthodes de
l'ECD. Mais il restera toujours à l'utilisateur de bien fixer ses choix
pour telle ou telle méthode en fonction de ses objectifs et de la nature
des données qu'il se propose de fouiller. Le récapitulatif donne
aussi un aperçu global sur l'utilisation des différentes
méthodes de fouille de données par tâche, mais notons quand
même que ce n'est pas un tableau exhaustif, il résume les
techniques les plus utilisées et qui apparaissent souvent dans la
littérature relative au domaine que nous avons abordé. Nous avons
aussi établi un tableau comparatif (voir Tableau 1.6) qui
mentionne les principaux avantages et inconvénients des
différentes tâches de l'ECD.
Chapitre I : L'extraction de connaissances à
partir de données biologiques - 28 -
|
Domaines d'utilisation
|
Méthodes de FDD
|
|
H M M
|
S V M
|
K - Moyenne
|
K - ppv
|
Réseaux de neurones
|
Arbre de
décision
|
Réseaux Bayésiens
|
Induction
de règles
|
Treillis
|
Autres
méthodes
|
|
PROTEOMIQUE
|
Identification de sequences
|
|
v
|
|
|
v
|
v
|
v
|
|
|
|
|
Comparaison de séquences
|
|
|
|
|
|
|
|
|
|
Alignement , similitude
|
|
Segmentation de sequences
|
v
|
|
|
|
|
|
|
|
|
|
|
Classification
|
v
|
|
|
|
|
|
|
|
|
n-gramme.
|
|
Prédiction structure
|
|
|
|
v
|
v
|
|
|
|
|
|
|
Prédiction domaine
|
|
|
|
|
v
|
|
|
|
|
|
|
Recherche de motifs fréquents
|
v
|
|
|
|
|
|
|
|
|
|
|
Recherche d'aberration
|
v
|
|
|
|
|
|
|
|
|
|
|
Recherche séquence exogène
|
v
|
|
|
|
|
|
|
|
|
|
|
Recherché
hétérogénéité
|
v
|
|
|
|
|
|
|
|
|
|
|
GENOMIQUE
|
Identification groupe gene
|
v
|
v
|
v
|
|
v
|
|
|
|
|
|
|
Expression de gènes
|
|
v
|
v
|
v
|
v
|
|
|
|
|
|
|
Identification de gènes
|
v
|
|
|
|
|
|
|
|
|
|
|
Prediction de gène
|
v
|
|
|
|
|
|
|
v
|
|
|
|
Comparaison de séquences
|
|
|
|
|
|
|
|
|
|
similarité
|
|
Groupement
|
v
|
|
|
|
|
|
|
|
v
|
|
|
COHORTE
|
Recherche de règles
d'association
|
v
|
|
|
|
|
|
v
|
v
|
|
Motifs frequents
|
|
Recherche de gènes candidats
|
v
|
|
|
|
|
|
|
|
|
Marqueurs génétiques
|
|
Fouille de données médicales
|
|
v
|
|
v
|
v
|
v
|
|
v
|
|
|
|
MALADIE
|
Interaction gène / environnement
|
|
|
|
|
|
|
v
|
v
|
|
|
|
Recherche gène candidat
|
v
|
|
|
|
|
|
|
|
|
|
|
Recherche cause
|
|
v
|
|
|
v
|
|
v
|
|
|
|
Tableau 1.4 : Les méthodes de FDD
utilisées en ECD biologiques.
Chapitre I : L'extraction de connaissances à
partir de données biologiques - 29 -
|
Tâches de
l'ECD
|
Types
|
Méthodes de FDD
|
|
Descriptive
|
Prédictive
|
Supervisée
|
Non Superv.
|
AED
|
H M M
|
Régression. linéaire.
|
S V M
|
K - Moyenne
|
K - ppv
|
Réseaux de neurones
|
Arbre de
décision
|
Réseaux
bayésiens
|
Algorithmes génétiques
|
Induction de règles
|
Treillis
|
|
Description
|
v
|
|
v
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Estimation
|
|
v
|
v
|
|
|
|
v
|
|
|
|
v
|
|
|
|
|
|
|
Prédiction
|
|
v
|
v
|
|
|
v
|
v
|
|
|
|
v
|
|
|
|
|
|
|
Classification
|
|
v
|
v
|
|
|
v
|
v
|
v
|
|
v
|
v
|
v
|
v
|
v
|
v
|
v
|
|
Groupement
|
v
|
|
|
v
|
|
|
|
|
v
|
v
|
v
|
|
|
|
|
|
|
Recherche Association
|
|
v
|
|
v
|
|
|
|
|
|
|
|
|
v
|
v
|
v
|
|
Tableau 1.5 : Les tâches et méthodes
utilisées en ECD.
|
Tâches de
l'ECD
|
Caractéristiques / Objectifs
|
Apprentissage
|
Avantages
|
Inconvénients
|
Algorithmes utilisés
|
|
Description
|
· Il s'agit de décrire les données
pour
essayer de découvrir et de
comprendre le
processus qui est à l'origine de ces données.
|
supervisé
|
Facile à mettre en oeuvre.
|
Difficiles à évaluer en cas de beaucoup de
variables.
|
· Stat. élémentaire ;
· Histogramme ;
· moy, écart-type ;
· ACP....
|
Estimation
|
· Consiste à estimer la valeur
d'une
variable à valeurs continues à partir des valeurs
d'autres attributs.
|
supervisé
|
En cas d'estimation numérique le résultat
est numérique.
|
Besoin d'un ensemble d'exemples
|
· Régression ;
· Réseaux de neurones ;
· Kppv.
|
Prédiction
|
· Consiste à prédire la valeur
future
d'un attribut en fonction d'autres attributs ;
· Se base sur le présent pour
trouver
des résultats dans le futur ;
· Assimilable à l'estimation mais
les
objets sont classés en fonction d'un comportement futur
prédit.
|
supervisé
|
Nécessite un tas de données assez
énorme pour pouvoir faire une prédiction assez
précise.
|
On ne peut vérifier la précision du
résultat tout de suite.
|
· Arbre de décision ;
· Réseaux de neurones ;
· Réseaux bayesiens.
|
Classification
|
· Consiste à examiner les
caractéristiques d'un objet et lui
attribuer
une classe ;
· Les classes sont connues à
l'avance
avec des profils particuliers.
|
supervisé
|
Robustesse (bruit, données manquantes,...)
;
Interprétabilité simple.
|
Taux d'erreur non négligeable ;
Temps d'exécution (construction, utilisation)
conséquent.
|
· Kppv ;
· Arbre de décision ;
· Réseaux de neurones ;
· Algo. Génétique ;
· HMM.
|
Groupement
|
· Il s'agit de grouper des objets en
se
basant sur leurs similarités ;
· Les objets sont les plus
similaires
possibles dans un groupe et moins
similaires possibles entre deux
groupes ;
· La similarité peut être
calculée pour
différents types de données. Elle
dépend des données utilisées et du type de
similarité recherchée.
|
non supervisé
|
Peut traiter # types de
données ;
Peut traiter les données bruitées et
isolées ;
Peut découvrir des clusters de # formes
;
Pour les attributs numériques, la distance est
bien définie.
|
L'interprétation des groupes identifiés
est plus difficile que la classification car les groupes ne sont pas connus
à l'avance ;
Pour les attributs énumératifs ou mixtes
la distance est difficile à définir.
|
· K moyennes ;
· Réseaux de neurones.
|
Recherche d'association
|
· Déterminer les attributs qui
sont
corrélés, i.e. découvrir des relations plus fines
entre les données.
|
non supervisée
|
Itemsets de tailles variables ;
Résultats clairs.
|
Produit énormément de règles
;
Nécessite un post-
traitement
humain.
|
· Apriori ;
· AIS ;
· FP-Growth.
|
|
Tableau 1.6 : Tableau comparatif des tâches
de l'ECD.
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
30 - I.7 Discussion sur l'ECD Biologiques
L'ECD est un domaine qui a connu une émergence
remarquable pendant la dernière décennie. Ce succès s'est
réalisé sur le champ scientifique et s'est prolongé au
champ commercial. En effet, plusieurs éditeurs d'outils de fouille de
données ont formé un marché riche de logiciels fiables
implémentant pratiquement la totalité des méthodes de la
littérature et ciblant toutes les tâches de fouille de
données. On trouve alors : WEKA et TANAGRA.
Ceci dit, l'ECD n'est pas du tout une clé avec
laquelle on fait jaillir les connaissances. Un outil de l'ECD n'est pas aussi
une boîte noire qu'on utilise ou un système totalement
automatique, son caractère interactif nécessite l'intervention
d'un humain et préférablement une personne ayant une expertise
dans son domaine d'application. Bien que l'ECD ait aidé à
répondre à plusieurs questions dans plusieurs domaines, il reste
un travail considérable à réaliser. Cet immense travail de
recherche, couramment présenté dans la littérature, montre
que l'ECD est un champ en cours d'émergence et qu'il reste quelques
défis que la communauté de fouille de données essaye de
relever et en particulier au niveau de l'ECD biologique. Nous citons quelques
uns :
Le traitement des données
complexes
En effet, la plupart des recherches dans le domaine
de l'ECD se focalisent sur l'extraction de connaissances à partir de
données simples, souvent sous la forme d'une table dont les colonnes
correspondent aux variables (items) et les lignes correspondent aux
entités décrites. Cependant, les banques de données
biologiques contiennent des données de différents formats et de
plus en plus complexes et non structurées telles que les données
textuelles et graphiques. Bien que des outils d'extraction de connaissances
à partir de textes (texte Mining), d'images (Image Mining) ont vu le
jour, ils ne semblent pas être aussi évolués pour permettre
l'extraction aisée de connaissances à partir de données
biologiques telles quelles se présentent aujourd'hui. Des travaux
novateurs ont été réalisés [Bahar et Chen, 2004],
[Abdelouhab et Atmani., 2008], [Benamina et Atmani, 2008], mais beaucoup reste
à faire.
Tenir compte des connaissances à
priori
Les systèmes d'ECD devraient permettre
à l'utilisateur d'exploiter ses connaissances à priori, non
seulement dans la sélection des données, mais aussi dans la phase
de fouille de données, car plus cette phase est guidée, plus les
connaissances dégagées
Chapitre I : L'extraction de connaissances
à partir de données biologiques -
31 -
seraient potentiellement utiles, et cet aspect
revêt une importance particulière en biologie parce que l'expert
capitalise une expertise qui peut très bien guider le processus de
fouille de données.
Optimisation
À cause de la très grande taille des
données (séquences génomiques, etc..), les algorithmes de
fouille de données devraient être performants. Le temps
d'exécution doit être pris en considération pour être
plus ou moins acceptable.
Variabilité des
données
Une particularité des données
biologiques, c'est qu'elles se trouvent sous divers formats définis par
les biologistes pour leurs besoins spécifiques. Ceci fait que du point
de vue traitements automatiques, nous nous retrouvons en face de divers formats
complexes et divers types de données : fichier texte, images, puce
à ADN, etc. Ceci, pose la question de la normalisation des formats des
données biologiques.
Evaluation des motifs
L'extraction de motifs dans des données de
grandes tailles (séquence biologique), donne généralement
un nombre très élevé de motifs. Peut être qu'il
faudra trouver des mesures du domaine de la biologie (en plus du support et de
la confiance) qui permettent l'évaluation des motifs pour dégager
ceux qui sont réellement intéressants.
I.8 Conclusion
Il est évident que l'ECD biologiques soit un
processus complexe. Cette complexité est induite par les formats de
données qui ne sont pas souvent les formats standards connus usuellement
en informatique. De plus cette complexité se traduit au niveau de
l'implémentation des différentes méthodes utilisées
: HMM, réseaux de neurones, etc. Ce qui rend la fouille de
données assez couteuse en particulier pour certaines méthodes
telle que la recherche d'association. De plus, les travaux de fouille en
biologie sont assez variés : prédiction de gènes,
recherche de motifs fréquents, etc. Peu de travaux sont consacrés
à l'optimisation des méthodes de fouille de données dans
un contexte biologique, et ce pour la spécificité des
données utilisées.
Notre travail porte précisément sur
l'extraction des règles d'association dans un contexte biologique, avec
post-traitement des résultats par optimisation par la machine
cellulaire, dont l'efficacité a été prouvée [Atmani
et Beldjilali, 2007], [Benamina et Atmani, 2008]. Le chapitre 2 est
consacré à l'extraction des règles d'association et
au
Chapitre I : L'extraction de connaissances à
partir de données biologiques - 32 -
processus utilisé pour les produire, et le
chapitre 3 étalera leurs modélisations booléennes par le
système BRI.
Chapitre II : L'extraction de règles d'association -
33 -
Chapitre II.
L'extraction de règles d'association
L'extraction des règles d'association est une
méthode descriptive de fouille de données qui a reçu
beaucoup d'intérêt de la part des chercheurs. On peut la
définir comme étant la recherche de relations entre des Items
dans un ensemble de données.
Cette technique est très utilisée pour
l'analyse des paniers. Il s'agit d'analyser l'ensemble des achats
effectués par les clients d'une entreprise commerciale. Chaque achat
contient un ensemble d'Items (articles), il correspond à un panier. Pour
cela, l'entreprise doit archiver les achats (transactions) effectués
dans une base de données binaire (Transaction, Items) où
l'attribut Transaction est une clé et Items est l'ensemble de n articles
article1, article2, ....articlen .
Plusieurs algorithmes d'extraction de règles
d'association à partir de bases de données ont été
proposés [Agrawal et al., 1993], [Agrawal et Srikant., 1994], [Savasere
et al., 1995], [Hip et al., 2000].
Motivations
L'ECD est un domaine relativement récent.
Plusieurs recherches intéressantes ont été faites et ont
produit des résultats satisfaisants. Il s'agit de techniques et de
méthodes nouvelles qui concernent chacune des phases du processus d'ECD,
en particulier la fouille de données. Il existe alors plusieurs travaux
développés pour l'extraction de motifs et particulièrement
les règles d'association. Dans la section qui suit, nous
présentons l'extraction de ce type de motifs et nous expliquons nos
choix, mais bien avant et pour la clarté nous donnons quelques
définitions.
Les Itemsets et les règles d'association.
Ils sont le produit du processus d'extraction des
règles d'association et ils tirent leurs origines des Items.
Définition 1 : Item
Un Item est un article au sens où il est pris en
extraction de règles d'association.
Définition 2 :
Itemset
Un Itemset est un ensemble de n Items noté :
{A, B, C, D....}. L'ensemble de tous les Itemsets possiblement formés
par les éléments d'Items est 2n.
Chapitre II : L'extraction de règles d'association -
34 -
Définition 3 : le support
Le support d'un Itemset k dans une base de
données, est l'ensemble de toutes les transactions qui supportent k, il
est noté supp(k).
Définition 4 : la confiance
La confiance dans une règle, notée
conf(X?Y) est la probabilité qu'une transaction supportant X supporte
également Y.
Exemple :
Etant donné une base de données, chaque
instance est un ensemble d'Items:
Transaction
|
Item
|
1
|
A C T W
|
2
|
C D W
|
3
|
A C T W
|
4
|
A C D W
|
5
|
A C D T W
|
6 C D T
|
|
Support
|
Items Set
|
100 % (6)
|
C
|
85
|
%
|
(5)
|
CW
|
67
|
%
|
(4)
|
A, D, T, AC, AW,CD, CT, ACW
|
50
|
%
|
(3)
|
AT, DW, TW, ACT, ATW, DW, CTW, ACTW,CDW
|
|
Item sets Frequent Maximum: ACTW, CDW
· Sous-Itemsets : CDW (3), CD (4), CW (5), DW (3),
C (6), D(4), W (5)
· CD W, conf = 3/4
|
=
|
75%
|
· CW D, conf = 3/5
|
=
|
60%
|
· DW = C, conf = 3/3
|
=
|
100%
|
· C DW, conf = 3/6
|
=
|
50%
|
· D = CW, conf = 3/4
|
=
|
75%
|
· W = CD, conf = 3/5
|
|
=
|
60%
|
|
II.1 Les règles d'association
Une règle d'association est une relation
d'implication X - Y entre deux ensembles d'Items X et Y tel
que XnY =Ø et X?Ø. X est appelé corps de la règle
et Y est la tête. Cette règle indique que les transactions qui
contiennent les articles de l'ensemble X ont tendance à contenir les
articles de l'ensemble Y. X est appelé condition ou prémisse et Y
est appelé résultat ou conclusion. Ces règles sont de la
forme :
Si {Item 1, Item 2, ..., Item j } Alors { Item
k,....,Item p }
Exemple : Si {gene1 = "1"} Alors {gene5="1"}
Cette règle est interprétée de la
manière suivante :
"Si la séquence possède gene1 Alors elle
possède gene5"
Chapitre II : L'extraction de règles d'association -
35 -
Les règles d'association induisent deux notions
:
- le support qui est le pourcentage (%) d'instances de la
base vérifiant la règle ou fréquence d'apparition ensemble
de la partie gauche et la partie droite de la règle. Support(X
-* Y) = p(XuY) = support ({X,Y})
- la confiance qui est la probabilité que la
partie droite de la règle soit vérifiée, si partie gauche
de la règle est vérifiée.
Confiance(X -* Y) = p(Y|X) = p(XuY) /
p(X) = support({X,Y}) / support({X})
II.2 L'induction et l'évaluation des
règles
L'induction est en effet l'extraction de règles
qui satisfont des seuils minimums prédéfinis de support et de
confiance à partir d'une base de données volumineuse [Agrawal et
Srikant., 1994]. Cette extraction se déroule en deux phases
:
(1) extraction des Itemsets fréquents
;
(2) génération des règles
d'association confiantes.
Le nombre d'Itemsets possibles pour une base de
données stockant n Items est 2n. Ceci rend le nombre de
règles encore plus énorme. Il en découle que l'extraction
de la totalité des Itemsets (pour ensuite tester leurs
fréquences) soit une opération très coûteuse voir
impossible. Les deux mesures qui évaluent le degré
d'intérêt de l'utilisateur envers un motif sont : le support et la
confiance. L'utilisateur mentionne alors un seuil minimal de support et un
seuil minimal de confiance, et les règles dont les confiances sont
supérieures au seuil fixé sont acceptées et les autres
sont alors rejetées. On peut dire alors que ces deux seuils sont des
contraintes d'extraction de règles. Ainsi, le problème
d'extraction de règles d'association est décomposé en deux
étapes :
- la première consiste à dégager
les Itemsets fréquents, i.e., les Itemsets dont le nombre d'occurrences
est supérieur au seuil minimum de support ;
- la seconde consiste à générer
des règles d'association à partir de ces Itemsets
fréquents qui respectent un seuil minimum de confiance. Cette seconde
étape est beaucoup plus simple. En effet, une fois les Itemsets
fréquents trouvés, il y a génération des
règles possibles pour chaque Itemset.
Génération des règles
La génération des règles est donc
une opération de transformation des ensembles d'Items en règles
de manière efficace. De ce fait, à partir d'un ensemble de
n Items, on
Chapitre II : L'extraction de règles d'association -
36 -
peut générer 2n-1
règles potentielles, et on ne gardera que les règles avec une
confiance supérieure au minimum fixé par
l'utilisateur.
On peut alors dire : Etant donné deux seuils
minimaux, s de support et c de confiance, l'extraction de règles
d'association se fait en deux étapes :
(1) Extraction des Itemsets fréquents (dont la
fréquence est supérieure à s)
(2) Extraction des règles d'associations
confiantes (dont la confiance est supérieure à c) à partir
des Itemsets fréquents.
Ce traitement se traduit par :
(1) un calcul coûteux des supports;
(2) une génération coûteuse des
règles ;
(3) un calcul coûteux de la confiance
;
(4) un parcours des données initiales
récurrent.
Évaluation des règles
La méthode d'extraction de règles
d'association peut produire des règles d'association triviales ou
inutiles. Ces règles triviales sont des règles évidentes
(par exemple : Si marié Alors non-célibataire) qui n'apporte pas
d'information supplémentaire. Les règles inutiles sont des
règles difficiles à interpréter qui peuvent provenir de
particularités propres à la liste des t-uples ayant servi
à l'apprentissage. De plus, une règle d'association est
destinée à être utilisée par la suite dans un but
décisionnel, il est nécessaire d'évaluer sa
qualité. Il faut qu'à l'étape de fouille, il y ait
suffisamment d'exemples vérifiant cette règle, et une
quantité de contre-exemples qui ne porte pas préjudice au sens
que prend cette règle dans son contexte d'extraction.
De nombreux critères d'évaluation
existent [Hip et al., 2000]. Ils peuvent être aussi subjectifs. L'expert
du domaine sait quels attributs il souhaite avoir dans les règles
d'association, mais ce cas de figure est très rare, car contraire au but
de l'ECD qui est la découverte des connaissances dont on ignore à
priori l'existence. Il existe également de nombreux critères
beaucoup plus objectifs qui consistent à étudier le nombre
d'exemples et de contre-exemples afin que la règle obtenue ne soit pas
trop générale ou évidente, car dans ce cas il n'y a rien
de nouveau. Les statistiques sont utilisées pour associer à
chaque règle d'association de la forme X ? Y des
mesures permettant d'avoir une idée de sa qualité :
- le support est une mesure dite d'utilité.
C'est la probabilité p (X, Y) pour que X et Y soient vrais en même
temps ;
Chapitre II : L'extraction de règles d'association -
37 -
- la confiance est une mesure dite de
précision. C'est la probabilité pour que Y soit vrai lorsque l'on
a X vrai, soit la probabilité conditionnelle p (Y | X).
Suivant le contexte dans lequel est extraite cette
règle, le taux d'erreur admissible dépend du facteur risque. Par
exemple, on exigera une qualité de cette règle d'association
beaucoup plus importante dans le domaine médical. Dans ce cas, les
valeurs des seuils minimaux de support et de confiance seront
élevées afin de ne retenir que les règles les plus fiables
dans le sens où elles ont très peu de contre-exemples (dont
l'existence peut être dangereuse). De nombreux autres indices de mesure
de la qualité d'une règle d'association existent [Agrawal et al.,
1993].
II.3 Les algorithmes d'extraction des règles
d'association
Dans cette section, nous essayons de présenter
quelques algorithmes d'extraction de règles association les plus
utilisés. Ils sont classés selon [Hip et al., 2000]:
· la stratégie de parcours de l'espace de
recherche ;
· la stratégie de calcul des supports des
Itemsets.
AIS [Agrawal et al., 1993],[Agrawal et
Srikant., 1994]
Cet algorithme fut le premier proposé pour
l'extraction de règles d'association. Il extrait des règles
d'association dont la tête est formée par un seul Item. Une
règle est alors de la forme X?Y avec Y un
élément de Items. Pour cela, la base de données est
parcourue plusieurs fois afin de dégager les Itemsets fréquents.
Le kème parcours est réalisé pour extraire les
Itemsets de cardinalité k qu'on appellera dorénavant k-Itemsets.
Pendant le premier parcours, c'est l'extraction des 1-Itemsets fréquents
(F1) qui se fait. Pour extraire les 2-Itemsets fréquents (F2),
l'algorithme parcourt la base de données pour tester les supports des
2-Itemsets appartenant à l'ensemble des 2-Itemsets candidats noté
C2. C2 est généré à partir de F1, ensemble des
1-Itemsets fréquents. Les 2-Itemsets candidats sont
générés en étendant les 1-Itemsets fréquents
par un Item de la même transaction. Pour généraliser, cet
algorithme parcourt deux fois la base de données pour
générer les k-Itemsets fréquents :
- la première fois sert à
générer les k-Itemsets candidats ;
- la deuxième fois sert à calculer le
support de chaque candidat, le candidat ayant un support supérieur au
seuil minimum est inséré dans l'ensemble Fk, sinon il est
rejeté.
Chapitre II : L'extraction de règles d'association -
38 -
L'inconvénient de l'algorithme AIS est le fait
qu'il scanne la base de données plusieurs fois et qu'il
génère trop de candidats.
Algorithme : AIS Début
Input : L1 = { large 1-Itemsets
}
C1= data base D
min_sup min_conf
Output : association rules R
L1 = { large 1-Itemsets }
for ( k=2 ;
Lk-1 ? Ø ; k++)
faire
ct=Ø
Forall transaction t E D
do begin
Lt=subset(Lk-1 , t)
forall large itemsets lt
E Lt do begin
if ( c E Ck ) then
add 1 to the count of c inthe corresponding entry in
Ck
else
add 1 to Ck with a count of
1
end
end
Lk={c E Ck / c.support =
min_sup}
end
L = Uk Lk ;
R=Generate_Rules(L) Fin
FP-Growth [Han et al., 2000]
C'est un algorithme qui réalise la tâche
après deux parcours de la base de données en s'appuyant sur une
structure d'arbre.
Le processus d'extraction de patterns se déroule
en deux grandes étapes :
(1) la construction de l'arbre appelée FP-Tree
(une représentation condensée de la base de données)
;
(2) la génération des patterns
fréquents à partir de cette structure, l'arbre FP-Tree. La racine
de l'arbre ne porte aucune information (`null'), alors qu'un noeud autre que la
racine porte deux informations, l'Item qu'il représente et son support.
Un index est associé à l'arbre FP-Tree, et il contient tous les
Items fréquents. A chaque Item est associé un pointeur vers le
premier noeud de l'arbre qui le contient.
Chapitre II : L'extraction de règles d'association -
39 -
La première étape qui consiste à
construire l'arbre FP-Tree, se déroule comme suit :
(a) la base de données est parcourue pour
calculer le support des Items. Les Items fréquents sont
générés dans un ensemble qu'on notera F1 ;
(b) la base de données est parcourue une
deuxième fois, et chaque transaction (ensemble d'Items) est triée
selon le support des Items de manière décroissante. Chaque
transaction t est représentée par sa tête p (l'Item le plus
fréquent dans ce panier) et par la queue Q (ensemble trié des
Items moins fréquents que p). Pendant le même parcours, pour
chaque transaction, une fonction Insert(p,Q,T) procède ainsi : Si la
racine T a un noeud fils N tel que N.Item=t.p alors son support est
incrémenté, sinon un noeud fils N est créé avec un
support égal à 1. Ensuite, on affecte récursivement
à p le premier Item de Q, et à Q le reste des Items, on
vérifie s'il existe un noeud N' fils de N tel que N'.Item=t.p, si oui on
incrémente son support, sinon on créé un nouveau noeud N'
(fils de N) avec un support égal à 1. Cette fonction est
exécutée récursivement jusqu'à ce que Q soit
vide.
Il faut noter que l'objectif de tri des Items (dans
l'ordre décroissant de leur fréquence) est de réduire la
taille de l'arbre, ainsi les Items les plus fréquents seraient
partagés par les transactions.
Algorithme : FP-Growth Début
Entrée : L'arbre FP-Tree noté T
Seuil minimum de support min_s
Sortie : Ensemble des Itemsets Fréquents :
F(D,min_s)
FP-Growth (T, á)
Si T contient un seul chemin Alors
Pour chaque combinaison f3 des noeuds du chemin
faire
Générer le pattern f3 U á avec
support=support minimum des noeuds dans f3
FinPour
Sinon
Pour chaque ai de l'index de T faire
Générer le pattern f3 = ai U á avec
support = ai.support
Construire le pattern de base de f3
Construire le FP-Tree conditionnel de f3 et l'affecter
à T
FinPour
Si T ? Ø Alors FP-Growth (T,f3)
Fin
Chapitre II : L'extraction de règles d'association -
40 -
Apriori [Agrawal et Srikant., 1994]
Cet algorithme fut une évolution dans
l'histoire de l'extraction des règles d'association. Il fonctionne selon
le principe suivant :
(1) générer les Itemsets ;
(2) calculer les fréquences des Itemsets
générés;
(3) garder les Itemsets qui ont un support minimum i.e.
les Itemsets fréquents ;
(4) générer et ne garder que les
règles avec une confiance minimale.
Apriori est plus efficient que AIS au niveau de la
génération des Itemsets candidats, du moment où il
élague encore plus l'espace de recherche. Le dégagement des
Itemsets fréquents se fait sur deux étapes : tout d'abord, les
Itemsets candidats sont générés, ensuite la base de
données est parcourue pour calculer les supports des candidats, et
accepter les candidats dont le support est supérieur au seuil minimum
comme étant des Itemsets fréquents. L'apport d'Apriori
réside dans la génération des candidats. En effet, un
k-Itemset candidat n'est généré que si tous ses
sous-ensembles de cardinal k-1 sont fréquents. Cependant, Apriori
«s'étouffe» sans aucun doute sur deux phases : la
génération des candidats qui est un processus complexe, et le
calcul des supports des Itemsets candidats qui nécessite plusieurs
parcours de la base de données.
Algorithme : Apriori Début
Entrée : Base de données D
Seuil minimum de support min_s Seuil minimum de confiance
min_c
Sortie : Ensemble des règles d'association
R
F1 = 1-Itemsets fréquents;
Pour ( k=2; Fk-1?Ø; k++ )
faire
Ck =Apriori_gen(Fk-1);
Pour ( chaque transaction t de D ) faire
Pour ( chaque candidat c de Ck ) faire Si ( c est inclus
dans t ) alors c.support++ ;
Finsi
Finpour
Finpour
Fk={c?Ck / c.support = min_s}
;
Finpour
F = Uk Fk ;
R=Generer_Regles(F) ;
Fin
Chapitre II : L'extraction de règles d'association -
41 -
Discussion
Nous avons consulté des articles dont l'objet
est l'étude comparative des algorithmes d'extraction de motifs
fréquents [Goethals et Van den Bussche, 2002], [Zaki et al., 1997], [Hip
et al., 2000]. Il est dit que l'application de ces algorithmes sur une
même base de données avec les mêmes seuils de support et de
confiance, donne globalement les mêmes résultats (les mêmes
règles d'association). Ceci est dû au fait que les algorithmes
d'extraction de règles d'association sont tous plus ou moins proche l'un
de l'autre. Il en découle que la comparaison concerne seulement les
performances des algorithmes et non la qualité des
résultats.
Nous avons constaté que ces études sont
subjectives car les conclusions tirées ne sont pas les mêmes.
Cette subjectivité est due au fait que le même algorithme peut
être implémenté de plusieurs manières
différentes d'où la différence de point de vue performance
et efficience.
Ceci dit, nous présentons ici quelques
conclusions se rapportant à Apriori communément tirées par
les études que nous avons consultées, ceci du fait que cet
algorithme est communément jugé plus ou moins efficace par
rapport aux autres :
· Apriori parcourt la base de données k
fois, k étant le cardinal du plus long Itemset fréquent. Ceci
entraîne beaucoup d'opérations d'entrée/sortie qui sont
coûteuses. Il faut noter aussi que dans le cas de bases de données
volumineuses, Apriori met du temps pour générer les Itemsets
fréquents. En effet dans ce type de base de données, le nombre
d'Itemsets fréquents est très grand, ce qui augmente le
coût de la génération des Itemsets candidats et du calcul
de leurs supports. En général, lorsque le nombre d'Itemsets
fréquents est grand, le coût d'extraction est élevé.
Le nombre d'Itemsets fréquents dépend du seuil minimum de support
(plus le seuil est petit, plus le nombre d'Itemset fréquent est grand)
et du volume de la base de données ;
· Apriori est l'algorithme le plus
approprié à l'exploration des bases de données. Sa
puissance réside dans son élagage, plus le minimum support est
petit plus l'élagage est faible.
Chapitre II : L'extraction de règles d'association -
42 -
II.4 Conclusion
Le processus de l'ECD biologiques adopté par
notre système est composé de 7 étapes majeures
:
1ere étape .
· Sélection et
prétraitement des données
A partir de la banque de donnée
NCBI1, il y a récupération des données
biologiques relatives aux souches ciblées par notre étude sous
leurs formats originaux. Les souches ciblées sont ceux dont l'annotation
a été finie à savoir : Mt H37Rv, Mt CDC1551, Mt F11, Mt
H37Ra. Un nettoyage et une mise en forme sont effectués afin de
dégager des descripteurs « attributs » possibles.
2eme étape .
· Transformation des
données
La transformation des données du format
original vers un formalisme approprié est faite et suivra alors une
«binarisation».
3eme étape .
· Production et évaluation
des règles d'association
La recherche des Itemsets et des règles
d'association est faite par l'algorithme Apriori [Agrawal et Srikant., 1994]
avec calcul systématique du support et de la confiance pour ne retenir
que les règles ayant une confiance dépassant la valeur
fixée par l'utilisateur.
4eme étape .
· Transformation
Les règles d'association trouvées sont
transformées puis représentées selon un formalisme
transitoire aidant à la production d'un graphe d'induction.
Ainsi la règle d'association Ri se verra
traduite en une règle booléenne transitoire selon le principe
suivant :
( Ri , Antécédenti , Conséquenti ,
support , confiance )
( Rti , Prémissei ( Antécédenti ) ,
Conclusioni ( Conséquenti ) )
5eme étape .
· Production du graphe
d'induction
Un graphe d'induction est construit selon le principe
suivant : un sommet désigne un noeud sur lequel on fait un test, avec
les résultats possibles binaires ou à valeur
multiple.
1
http://www.ncbi.nlm.nih.gov/Database/
http://www.ncbi.nlm.nih.gov/genomes/genlist.cgi?taxid=2&type=0&name=Complete%20Bacteria
Chapitre II : L'extraction de règles d'association -
43 -
6eme étape : Production des règles
cellulaires
(1) Génération des règles
cellulaires : Elles sont déduites à partir du graphe d'induction
et auront la forme suivante:
Rck : Si { Prémissek } Alors { Conclusionk , Sommetk
}
où Prémissek
et Conclusionk sont composée
d'Items et Sommetk le noeud du graphe d'où la
règle est déduite.
(2) Représentation cellulaire : Les
règles générées auparavant (6.1) sont
représentées en couches cellulaires selon le principe cellulaire
(voir III.3). Schématiquement nous aurons :
{ Rck } REGLES et { Prémissek , Conclusionk , Sommetk
} FAITS
7eme étape : Intégration
La machine cellulaire intégrera et exploitera
la représentation cellulaire et les matrices d'E/S à travers une
inférence en chaînage avant pour enrichir la base de
connaissances.
Le chapitre suivant nous montrera le processus de
génération et d'intégration des règles
booléennes par le système BRI. Ce processus commence à
partir de l'étape 4, la transformation et fini à
l'intégration des résultats dans la base de
connaissances.
Chapitre III : Modélisation booléenne des
règles d'association - 44 -
Chapitre III.
Modélisation booléenne des règles
d'association
Les règles d'association aident le spécialiste
du domaine à interpréter ou expliquer certains
phénomènes biologiques liées à une pathologie
donnée, comme par exemple la cooccurrence de gènes liés.
Mais pour un système d'inférence, ces règles ne se
prêtent pas directement pour un stockage dans sa base de connaissance.
Pour cela, elles seront modélisées selon le principe
adopté par la machine cellulaire CASI [Atmani et Beldjilali, 2007], pour
réduire la complexité de stockage et le temps de réponse
lors de leurs utilisations.
Le système BRI développé adopte la
démarche suivante : transformation puis production de règles
booléennes inductives. Ainsi, les règles d'association sont
prétraitées et transformées en règles transitoires
nécessaires à la création d'un graphe d'induction qui
permettra de produire les règles booléennes inductives dans une
étape finale, et elles seront représentées en couches
cellulaires. Ainsi, BRI procède selon le principe
schématisé par la Figure 3.1 suivante :
Règles d'association
CIE (Cellular Inference Engine)
Règles transitoires
Graphe d'induction
Règles Booléennes
Figure 3.1: Le système BRI (Boolean Rule
Induction).
III.1 Le moteur d'inférence cellulaire :
architecture et principe de fonctionnement
Le module CIE (Cellular Inference Engine)
simule le fonctionnement du cycle de base d'un moteur d'inférence en
utilisant deux couches finies d'automates finis. La première couche,
CELFAIT, pour la base des faits, et la deuxième couche,
CELREGLE, pour la base de règles.
Chapitre III : Modélisation booléenne des
règles d'association - 45 -
Chaque cellule au temps t+1 ne dépend que de
l'état des ses voisines et du sien au temps t. Dans chaque
couche, le contenu d'une cellule détermine si et comment elle participe
à chaque étape d'inférence : à chaque étape,
une cellule peut être active (1) ou passive (0), c'est-à-dire
participe ou non à l'inférence.
Atmani et Beldjilali (2007) ont supposés qu'il y a
l cellules dans la couche CELFAIT, et r cellules
dans la couche CELREGLE. Toute cellule i de la
première couche CELFAIT est considérée comme fait
établi si sa valeur est 1, sinon, elle est considérée
comme fait à établir. Toute cellule j de la
deuxième couche CELREGLE est considérée comme une
règle candidate si sa valeur est 1, sinon, elle est
considérée comme une règle qui ne doit pas participer
à l'inférence.
Les états des cellules se composent de trois parties :
EF, IF et SF, respectivement ER, IR
et SR, sont l'entrée, l'état interne et la sortie
d'une cellule de CELFAIT, respectivement d'une cellule de
CELREGLE. L'état interne, IF d'une cellule de
CELFAIT indique le rôle du fait : dans le cas d'un graphe
d'induction IF = 0 correspond à un fait du type sommet (si),
IF = 1 correspond à un fait du type attribut=valeur
(Xi = valeur). Pour une cellule de CELREGLE, l'état
interne IR peut être utilisé comme coefficient de
probabilité que nous n'aborderons pas dans ce mémoire.
Pour illustrer l'architecture et le principe de fonctionnement
du module CIE, coeur du système BRI, nous
considérons la partie du graphe, extraite de l'article Benamina et
Atmani (2008), obtenue en utilisant les partitions So = {so}, S1 = {s1, s2} et
S2 = {s3, s4, s5} (voir Figure 3.2).
R1: Si so alors (X3 = Elevée) et s1
R2: Si so alors (X3 = Normale) et s2
R3: Si s1 alors (X1 = Soleil) et s3
R4: Si s1 alors (X1 = Couvert) et s4
R5: Si s1 alors (X1 = Pluie) et s5
Base de connaissances
Figure 3.2. Les partitions So, S1 et
S2.
Le Tableau 3.1 montre comment la base de connaissance
extraite à partir de ce graphe d'induction est représentée
par les couches CELFAIT et CELREGLE. Initialement, toutes les
entrées des cellules dans la couche CELFAIT sont passives
(EF = 0), exceptées celles qui représentent la base des
faits initiale (EF(1) = 1).
Les matrices d'incidence RE et Rs représentent la
relation entrée/sortie des Faits et sont utilisées en
chaînage avant. On peut également utiliser Rs comme
relation d'entrée et RE comme relation de
Chapitre III : Modélisation booléenne des
règles d'association - 46 -
sortie pour lancer une inférence en chaînage
arrière. Notez qu'aucune cellule du voisinage d'une cellule qui
appartient à CELFAIT (respectivement à
CELREGLE) n'appartient pas à la couche CELFAIT
(respectivement à CELREGLE).
R1
R2
R3
R4
R5
CELREGLE (Règles)
ER
0
0
0
0
0
IR
1
1
1
1
1
SR
1
1
1
1
1
CELFAIT( Faits)
|
|
EF
|
IF
|
SF
|
|
so
|
1
|
0
|
0
|
|
X3 = Elevée
|
0
|
1
|
0
|
|
s1
|
0
|
0
|
0
|
|
X3 = Normale
|
0
|
1
|
0
|
|
s2
|
0
|
0
|
0
|
|
X1 = Soleil
|
0
|
1
|
0
|
|
s3
|
0
|
0
|
0
|
|
X1 = Couvert
|
0
|
1
|
0
|
|
s4
|
0
|
0
|
0
|
|
X1 = Pluie
|
0
|
1
|
0
|
|
s5
|
0
|
0
|
0
|
|
|
Tableau 3.1 : Représentation Cellulaire de la base
de connaissances de la Figure 3.2.
Le voisinage est introduit par la notion de matrice
d'incidence. Dans le Tableau 3.2 sont respectivement
représentées les matrices d'incidence d'entrée RE
et de sortie Rs de l'automate cellulaire. La relation d'entrée,
notée iREj, est formulée comme suit : Vi E
[1,1], Vj E [1, r], si (le Fait i E à la
Prémisse de la règle j) alors RE(i, j) - 1. De
même la relation de sortie, notée iRsj, est
formulée comme suit : Vi E [1,1], Vj E [1, r], si (le
Fait i E à la Conclusion de la règle j) alors
Rs(i, j) - 1.
|
RE
|
R1
|
R2
|
R3
|
R4
|
R5
|
|
so
|
1
|
1
|
|
|
|
|
X3 = Elevée
|
|
|
|
|
|
|
s1
|
|
|
1
|
1
|
1
|
|
X3 = Normale
|
|
|
|
|
|
|
s2
|
|
|
|
|
|
|
X1 = Soleil
|
|
|
|
|
|
|
s3
|
|
|
|
|
|
|
X1 = Couvert
|
|
|
|
|
|
|
s4
|
|
|
|
|
|
|
X1 = Pluie
|
|
|
|
|
|
|
s5
|
|
|
|
|
|
|
Rs
|
R1
|
R2
|
R3
|
R4
|
R5
|
|
so
|
|
|
|
|
|
|
X3 = Elevée
|
1
|
|
|
|
|
|
s1
|
1
|
|
|
|
|
|
X3 = Normale
|
|
1
|
|
|
|
|
s2
|
|
1
|
|
|
|
|
X1 = Soleil
|
|
|
1
|
|
|
|
s3
|
|
|
1
|
|
|
|
X1 = Couvert
|
|
|
|
1
|
|
|
s4
|
|
|
|
1
|
|
|
X1 = Pluie
|
|
|
|
|
1
|
|
s5
|
|
|
|
|
1
|
Tableau 3.2 : Les matrices d'incidences d'Entrée RE
et de sortie Rs pour la Figure 3.2.
Pour définir la dynamique du CIE, nous allons
rappeler que le cycle de base d'un moteur d'inférence, pour
établir un fait F en chaînage avant, fonctionne
traditionnellement comme suit :
· Recherche des règles applicables
(évaluation et sélection) ;
· Choisir une parmi ces règles, par exemple R
(filtrage) ;
· Appliquer et ajouter la partie conclusion de R à
la base des faits (exécution).
Le cycle est répété jusqu'à ce
que le fait F soit ajouté à la base des faits, ou s'arrête
lorsqu'aucune règle n'est applicable.
Chapitre III : Modélisation booléenne
des règles d'association - 47 -
La dynamique de l'automate cellulaire CIE, est assurée
par deux fonctions de transitions ä5678 et ä9:;<,
où ä5678 correspond à la phase
d'évaluation, de sélection et de filtrage, et
ä9:;< correspond à la phase d'exécution.
· la fonction de transition =>?@A:
/EB, CB, SB, ER, CR, SR0
· la fonction de transition =OPQR:
|
DEFGH
IJJK /EB, CB, EB, ER L /R M N EB0, CR, SR0
|
|
/EB, CB, SB, ER, CR, SR0
|
DSTUV
IJJK /EB L /R! N ER0, CB, SB, ER, CR, ERWWWW
|
0
|
|
Où la matrice R M désigne la
transposé de la matrice R .
Nous considérons Xo la configuration initiale de
l'automate cellulaire et, que
Y = =OPQR° =>?@A la fonction de
transition globale : Y/X00 = X1si Xo
|
DEFGH DSTUV
IJJK X'~ et X'~ IJJK Xl
|
|
Supposons que X = {Xo, X1, ..., X[}
est l'ensemble des configurations de notre automate cellulaire.
L'évolution discrète de l'automate, d'une
génération à une autre, est définie par la
séquence Xo, X1, ..., X[,
où X1\1 = Y/X10.
III.2 La modélisation booléenne
Les règles d'association produites sont
transformées selon le principe suivant :
· les Items de Antécédent vont servir
à constituer la Prémisse de la règle ;
· les Items de Conséquent vont servir à
créer la Conclusion de la règle.
Les règles transitoires sont stockées dans une
base de données qui servira à produire le graphe d'induction
selon le principe suivant : un sommet désigne un noeud sur lequel on
fait un test avec les résultats possibles, binaires ou
multivalués. Ainsi, le graphe d'induction permettra de produire les
règles cellulaires ( Rc ) sous la forme :
Rci : Si Premissei Alors Conclusioni
Avec une représentation cellulaire selon le principe
suivant :
· les Items des Premissei et des Conclusioni
vont constituer les faits : CELFAIT ;
· les Rci vont constituer les règles :
CELREGLE.
Ces règles produites seront intégrées
dans la base de connaissance de CIE pour exploitation en inférence.
Chapitre III : Modélisation booléenne des
règles d'association - 48 -
|
Règle
|
Antécédent
|
Conséquent
|
Support %
|
Confiance %
|
|
R1
|
aceA-2=1
|
pstS-3=1
|
45
|
77
|
|
R2
|
pstS-3=1
|
Aacd=1
|
60
|
80
|
|
.....
|
....
|
.
|
|
|
Rt1 : Si aceA-2=1 Alors pstS-3=1
Rt2 : Si pstS-3=1 , aacd= 1 Alors rpsG=1, aroK=1 ....
Graphe d'induction
Rc1 : Si {s0} Alors {pstS-3=1, s1}
Rc2 : Si {s0} Alors {rpsG=1, aroK=1, s2}
......
CELFAIT
s0
pstS-3=0
CELREGLE
R1
|
FAITS
|
|
EF
|
IF
|
SF
|
|
1
|
0
|
0
|
|
0
|
1
|
0
|
|
|
|

|
REGLES
|
|
ER
|
IR
|
SR
|
|
0
|
1
|
1
|
|
...
|
|
|
RE
|
Rc1
|
Rc2
|
|
|
s0
|
1
|
1
|
|
|
pstS-3=1
|
0
|
0
|
|
|
....
|
...
|
...
|
|
RS
|
Rc1
|
Rc2
|
....
|
|
s0
|
0
|
0
|
|
|
pstS-3=1
|
1
|
0
|
|
|
...
|
...
|
...
|
Figure 3.3 : Illustration du principe d'induction des
règles booléennes
inductives par BRI.
III.3 Exemple d'illustration d'induction des
règles booléennes
Le processus général que notre système
d'apprentissage applique à un échantillon est illustré par
un exemple à partir de la 3eme étape du processus de
l'ECD adopté (Chapitre II-Section 4). Nous supposons avoir obtenu les 3
règles d'association suivantes, avec les gènes suivants : aceA-2,
asd, pstS-3, rpsG, aroK, argC, et phhB.
3eme étape : Production et évaluation des
règles d'association
|
Règle
|
Antécédent
|
Conséquent
|
Support %
|
Confiance %
|
|
R1
|
aceA-2=1
|
pstS-3=1
|
45 %
|
77 %
|
|
R2
|
asd=1
|
rpsG=1, aroK=1
|
80 %
|
90 %
|
|
R3
|
aceA-2=1, phhB=1
|
argC=1
|
45 %
|
70 %
|
|
FAITS
|
|
s0
|
|
pstS-3=1
|
|
s1
|
|
nul
|
|
s2
|
|
argG=1
|
|
s3
|
|
rpsG=1
|
|
aroK=1
|
|
S4
|
|
CELFAIT
|
|
EF
|
IF
|
SF
|
|
1
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
CELREGLE
|
|
ER
|
IR
|
SR
|
|
0
|
1
|
1
|
|
0
|
1
|
1
|
|
0
|
1
|
1
|
|
0
|
1
|
1
|
Chapitre III : Modélisation booléenne des
règles d'association - 49 -
4eme étape : Transformation
|
Règle
|
Prémisse
|
Conclusion
|
|
Rt1
|
aceA-2=1
|
pstS-3=1
|
|
Rt2
|
asd=1
|
rpsG=1, aroK=1
|
|
Rt3
|
aceA-2=1, phhB=1
|
argC=1
|
Seme étape : Production du graphe
d'induction
nul
pstS-3=1
s0
aceA-2
s2 s1
asd
phhB
argC=1
S4
rpsG=1, aroK=1
s3
6eme étape : Production des règles
cellulaires ( Rci )
O Génération des règles cellulaires
Rc1 : Si {s0 } Alors
{pstS-3=1, s1 }
Rc2 : Si {s0 } Alors {nul,
s2 }
Rc3 : Si {s1 } Alors
{argC=1, s3 }
Rc4 : Si {s2 } Alors {rpsG=1,
aroK=1, s4 }
O Représentation des règles cellulaires
Les règles cellulaires extraites à partir du graphe
(5eme étape) sont
représentées par les couches CELFAIT et CELREGLE et les matrices
d'E/S.
Les couches cellulaires de l'automate cellulaire
CASI
(1) La couche CELFAIT (2) La couche CELREGLE
REGLES
Rc1
Rc2
Rc3
Rc4
Chapitre III : Modélisation booléenne des
règles d'association - 50 -
(3) La matrice d'incidence d'entrée
RE(relation d'entrée)
|
RE
|
Rc1
|
Rc2
|
Rc3
|
Rc4
|
|
s0
|
1
|
1
|
0
|
0
|
|
pstS-3=1
|
0
|
0
|
0
|
0
|
|
s1
|
0
|
0
|
1
|
0
|
|
nul
|
0
|
0
|
0
|
0
|
|
s2
|
0
|
0
|
0
|
1
|
|
argC=1
|
0
|
0
|
0
|
0
|
|
s3
|
0
|
0
|
0
|
0
|
|
rpsG=1
|
0
|
0
|
0
|
0
|
|
aroK=1
|
0
|
0
|
0
|
0
|
|
S4
|
0
|
0
|
0
|
0
|
(4) La matrice d'incidence de sortie RS(relation de
sortie)
|
RS
|
Rc1
|
Rc2
|
Rc3
|
Rc4
|
|
s0
|
0
|
0
|
0
|
0
|
|
pstS-3=1
|
1
|
0
|
0
|
0
|
|
s1
|
1
|
0
|
0
|
0
|
|
nul
|
0
|
1
|
0
|
0
|
|
s2
|
0
|
1
|
0
|
0
|
|
argC=1
|
0
|
0
|
1
|
0
|
|
s3
|
0
|
0
|
1
|
0
|
|
rpsG=1
|
0
|
0
|
0
|
1
|
|
aroK=1
|
0
|
0
|
0
|
1
|
|
S4
|
0
|
0
|
0
|
1
|
|
Pour CELFAIT
A l'état initial :
|
|
|
|
|
Si cellule (i) base de faits initiale : fait déjà
établi
|
Alors
|
EF(i)=1.
|
|
Si
|
cellule (i) 0 base de faits initiale : fait à
établir
|
Alors
|
EF(i)=0.
|
|
Si
|
un fait est du type attribut = valeur
|
Alors
|
IF(i)=1.
|
|
Si
|
un fait est du type sommet
|
Alors
|
IF(i)=0.
|
Pour CELREGLE
Si cellule(i) =1 Alors Ri candidate i.e. participe à
l'inférence
Si cellule(i) =0 Alors Ri non candidate i.e. ne participe pas
à l'inférence Pour la matrice d'entrée RE :
Si faiti ? à Prémisse de Rj Alors RE(i,j)
=1
Pour la matrice de sortie RS :
Si faiti ? à Conclusion de Rj Alors RS(i,j) =1
III.4 La dynamique du moteur d'inférence
cellulaire
La dynamique de l'automate cellulaire CIE, pour simuler le
fonctionnement d'un Moteur d'Inférence, utilise deux fonctions de
transitions äfact et
ärule, où äfact
correspond à la phase d'évaluation, de sélection et de
filtrage, et ärule correspond à la phase
d'exécution.
(1) Évaluation, sélection et filtrage (application
de äfact )
(EF, IF, SF, ER, IR, SR)
|
DEFGH
IJJK (EF, IF, EF, ER + (Ri
· EF), IR, SR)
|
|
(2) Exécution (application de
ärule ) (EF, IF, SF, ER, IR, SR)
|
DSTUV
IJJK (EF + (Rs
· ER), IF, SF, ER, IR, ER
|
)
|
Chapitre III : Modélisation booléenne des
règles d'association - 51 -
Nous montrons une simulation sur CELFAIT et CELREGLE de l'exemple
illustré auparavant, en considérant que ^_ est la configuration
initiale de l'automate cellulaire
`_
REGLES
Rc1
Rc2
Rc3
Rc4
|
FAITS
|
|
s0
|
|
pstS-3=1
|
|
s1
|
|
nul
|
|
s2
|
|
argG=1
|
|
s3
|
|
rpsG=1
|
|
aroK=1
|
|
S4
|
|
CELFAIT
|
|
EF
|
IF
|
SF
|
|
1
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
CELREGLE
|
|
ER
|
IR
|
SR
|
|
0
|
1
|
1
|
|
0
|
1
|
1
|
|
0
|
1
|
1
|
|
0
|
1
|
1
|
|
(1) Application de : /EB, CB, SB, ER,
CR, SR0
|
DEFGH
IJJK /EB, CB, EB, ER L /R M N EB0, CR, SR0
|
REGLES
Rc1
Rc2
Rc3
Rc4
|
FAITS
|
|
s0
|
|
pstS-3=1
|
|
s1
|
|
nul
|
|
s2
|
|
argG=1
|
|
s3
|
|
rpsG=1
|
|
aroK=1
|
|
S4
|
|
CELFAIT
|
|
EF
|
IF
|
SF
|
|
1
|
0
|
1
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
CELREGLE
|
|
ER
|
IR
|
SR
|
|
1
|
1
|
1
|
|
1
|
1
|
1
|
|
0
|
1
|
1
|
|
0
|
1
|
1
|
DSTUV
(2) Application de : /EB, CB, SB, ER,
CR, SR0 IJJK /EB L /R! N ER0, CB, SB, ER, CR, ERWWWW0
`a
REGLES
Rc1
Rc2
Rc3
Rc4
|
FAITS
|
|
s0
|
|
pstS-3=1
|
|
s1
|
|
nul
|
|
s2
|
|
argG=1
|
|
s3
|
|
rpsG=1
|
|
aroK=1
|
|
S4
|
|
CELFAIT
|
|
EF
|
IF
|
SF
|
|
1
|
0
|
1
|
|
1
|
1
|
0
|
|
1
|
0
|
0
|
|
1
|
1
|
0
|
|
1
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
0
|
1
|
0
|
|
0
|
1
|
0
|
|
0
|
0
|
0
|
|
CELREGLE
|
|
ER
|
IR
|
SR
|
|
1
|
1
|
0
|
|
1
|
1
|
0
|
|
0
|
1
|
1
|
|
0
|
1
|
1
|
(3) Application de la fonction de transition
globale Y = =OPQR° =>?@A
DEFGH DSTUV
La machine cellulaire passe de ^a à ^b avec Y/X~0 ~ X si X
IJJK X'~ et X'~ IJJK X2
Chapitre III : Modélisation booléenne des
règles d'association - 52 -
REGLES
Rc1
Rc2
Rc3
Rc4
|
FAITS
|
|
s0
|
|
pstS-3=1
|
|
s1
|
|
nul
|
|
s2
|
|
argG=1
|
|
s3
|
|
rpsG=1
|
|
aroK=1
|
|
S4
|
|
CELFAIT
|
|
EF
|
IF
|
SF
|
|
1
|
0
|
1
|
|
1
|
1
|
1
|
|
1
|
0
|
1
|
|
1
|
1
|
1
|
|
1
|
0
|
1
|
|
1
|
1
|
0
|
|
1
|
0
|
0
|
|
1
|
1
|
0
|
|
1
|
1
|
0
|
|
1
|
0
|
0
|
|
CELREGLE
|
|
ER
|
IR
|
SR
|
|
1
|
1
|
0
|
|
1
|
1
|
0
|
|
1
|
1
|
0
|
|
1
|
1
|
0
|
Le cycle s'arrête car aucune
règle n'est applicable.
III.5 Conclusion
Deux
motivon ati s
concurrentes nous ont
amenés à adoper t le
principe des
automates celluslaire
pour les systèmes à
base de règles d'assoon.
ciati En effet, nous
avons non seun leme t
souhaité avoir une
base de règles
booléenne op, timale mais
nous avons égn aleme
t souhaité améliorer la
gestion des connaissances par le moteur
d'inn fére ce
cellulaire
CIE.
Qund a il s'agit
de l'induction boonn
lée e des règles
d'assoon, ciati nous
devons
impérativneme
t
passer par les
étapes suivantes
:
1) Importer dans BRI les règles
d'assoon ciati
produite par la
fouille de données ;
2) Transformer les règles d'assoon
ciati importées en règles
transitoires selon un format
simplifié aidant à l'étape suivante ;
3) Produire un graphe, à partir
des règles
transitoires , qui
permet de générer des
règles cellulaires ;
4) Produire les règles
cellulaires à partir du graphe induit ;
5) Représenter ces règles
cellulaires selon l CELREGLE, RE,
RS).
|
e prinp ci e
de la machine CASI
|
(CELFAIT,
|
Les inférences du moteur CIE dans BRI se
résume comme suit :
· Initialisation
de la BC par automate
cellulaire : Cela revn ie t
à initialiser les couh c es
CELFAIT
et CELREGLE et générer
les matrices d'incidn e ces
et ;
·
Inférence des
règles en avn a t : C'est le
rôle du CIE ; Pour
déduire les faits buts le
module
d'inn fére ce
utilise les fonctions de
tranon siti et ;
Chapitre III : Modélisation booléenne
des règles d'association - 53 -
·
Inférence des
règles en arrière : C'est le rôle
du même CIE ; Pour induire les faits
hypothèses
le module
d'inférence utilise les mêmes
fonctions de
transition et mais en
permun ta t les
deux matrices
d'incidn e ces
d'entrée et de sortie.
Les avang ta es
de ce principe
boon lée basé
sur l'automate
cellulaire peuvn e t être récapu
it lés selon Benan mi a et
Atmani (200 8) comme suit
:
· La représenon
tati de la connn aissa ce
ainsi que son
conô tr le sont simp, les
sous forme de matrices
binaires exigeant
un prétraitement
minimal ;
· La facilité de
l'implémentation des
fonctions de tranon
siti et qui sont de
basse
compx, le ité
efficaces et robustes pour des valeurs extrêmes ;
· Les résultats
sont simples pour être
insérés et utilisés par
un système expert
;
· La matrice
d'incidence,
RE, facilite la transfoon rmati
de règles dans
des
expressions
équivn
ale tes boo léenn, es qui
nous permet
d'utiliser l'algb è
re de Boole élémentaire
pour examiner
d'autres
simplification.s
Chapitre IV : Conception et expérimentation du
système BIODM - 54 -
Chapitre IV.
Conception et expérimentation du système
BIODM
Une étude préalable des
caractéristiques des séquences biologiques était
nécessaire afin de comprendre les mécanismes régissant la
structure et le contenu des fichiers des séquences, et poser par la
suite des hypothèses de travail quant aux données
(séquences) utilisées. Cette étude a permis donc de
dégager des éléments importants, tels que les attributs,
les types de données utilisés, la taille, etc. Ces
éléments aident à structurer les données
nécessaires au futur système.
IV.1 Etude et choix des données biologiques pour
expérimentation
Nous avons fait une étude des
caractéristiques des séquences des souches du Mycobacterium
Tuberculosis à travers les gènes et protéines
associées. Cette étude a permis de cibler les données
à importer (voir Tableau 0.2), de la banque de données
NCBI, et qui pourraient réellement faire profiter notre
expérimentation. Pour cela, nous avons utilisé les souches
complètement annotées (voir Figure 0.2).
Ensuite, nous avons pris en considération non
seulement les gènes mais aussi les protéines. Il s'agit en fait
d'augmenter le gisement de données afin d'obtenir des connaissances
diversifiées et potentiellement utiles au spécialiste du
domaine.
1er aspect d'étude : les
gènes
La pathogénicité s'exprime par les
gènes responsables de l'invasion. Les gènes qui codent pour des
toxines impliquées dans les transferts horizontaux (gènes de
virulence), co-régulés, sur exprimés, ou sous
exprimés sont autant de cibles potentiels pour étudier cet
aspect. De ce fait, une étude de la structure du fichier des
gènes a été faite afin de savoir les nettoyer pour ne
garder que les données informatives et les exploiter, tels que le nom du
gène, son identifiant, etc.
2eme aspect d'étude : les
protéines
D'après la structure des protéines, il
est constaté que beaucoup s'organisent en domaines ayant une structure
et remplissant des fonctions diverses dont celles de la virulence ou la
résistance à certains antibiotiques. Une étude des
fichiers des protéines a été faite afin de
déterminer les données élémentaires à
prendre en considération dans notre processus d'ECD, à savoir le
nom de la protéine, son code, sa localisation, etc.
Chapitre IV : Conception et expérimentation du
système BIODM - 55 -
3eme aspect d'étude : les RDs et
MIRUs
La génomique comparative a montré
l'utilité des MIRUs en tant qu'éléments qui renseignent
beaucoup sur les souches bactériologiques, et aussi l'existence des RDs
« Région de Différence », qui sont présentes
chez une souche et absentes chez une autre, ces régions codent pour de
petites protéines qui appartiennent à des familles reconnues par
leurs virulences [Yokoyama et al., 2007]. Dès lors, ces familles de
protéines sont très intéressantes du point de vue
préventif (antigène protecteur) et thérapeutique (cibles
de médicaments). Une confirmation a été faite
récemment que les gènes de ces régions RDs pourraient
être impliqués dans la virulence du Mycobacterium Tuberculosis
[Ferdinand et al., 2004]. Donc, les données sur les RDs et MIRU sont
très intéressantes, néanmoins, elles sont
écartées de notre étude à cause de la
complexité de ces types de données, qu'il faudra étudier
en profondeur, et aussi de la non disponibilité des données
à l'état brut en quantités suffisantes dans les banques de
données biologiques.
IV.2 Architecture du système BIODM ( BIOlogical
Data Mining)
Notre système est composé de deux grands
modules, le premier produit des règles d'association et les transmet au
deuxième module (BRI) pour générer des règles
booléennes.
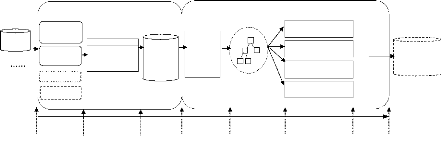
NCBI
Sélection, Prétraitement
Séquences génomiques
Séquences protéiques
RD
MIRU
Transformation Production
Evaluation.
Données structurées
Règles
d'assoc-
iation
Transformation Production Production de
Règles.
Transit-
oires
Graphe d'induction
Graphe d'induction
règles cellulaires
Boolean Rules Induction
CELRULE
CELFACT
REGLES
FAITS
Intégration
CASI
Knowledge
Base
Figure 4.1: Architecture du système
BIODM.
Chapitre IV : Conception et expérimentation du
système BIODM - 56 -
Le système BIODM fonctionne selon les 7
principales étapes suivantes :
1ere étape : Sélection et
prétraitement des données
Récupération des données
biologiques relatives aux souches concernées par notre étude,
sous leurs formats originaux, à partir de la banques de données
biologiques NCBI, avec nettoyage et mise en forme de celles-ci (voir
Tableau 0.2).
2eme étape :
Transformation des données
Transformation des données du format original vers
un formalisme base de données.
3eme étape : Production et évaluation
des règles d'association
Recherche des Itemsets et des règles
d'association par l'algorithme Apriori. Les règles d'association auront
le formalisme suivant :
( Ri , Antécédenti ,
Conséquenti , support , confiance)
4eme étape : Transformation des règles
d'association
Transformation des règles d'association selon
un formalisme transitoire aidant à la production d'un graphe
d'induction. Les règles transformées auront le
formalisme
suivant : ( Rti , Prémissei , Conclusioni
)
5eme étape : Production du graphe
d'induction
Production d'un graphe d'induction aidant à la
génération des règles booléennes
inductives.
6eme étape : Production des règles
cellulaires
Génération des règles cellulaires
( Rc ) à partir du graphe d'induction. Elles auront la forme
suivante : Rck : Si { Prémissek } Alors { Conclusionk , Sommetk
}
Elles seront représentées selon le
formalisme cellulaire. (Chapitre III-Section2).
7eme étape :
Intégration
Enfin, on intégrera les règles
générées dans la machine cellulaire CASI qui les
exploitera à travers une inférence en chaînage avant pour
enrichir sa base de connaissances.
Chapitre IV : Conception et expérimentation du
système BIODM - 57 -
IV.3 Le processus de l'ECD biologiques
Le processus de fouille de données
adopté par notre système suivra les 7 étapes majeures
définies auparavant:
1ère étape : Sélection et
prétraitement des données
A cette étape, nous récupérerons
les séquences génomiques et protéiques des souches
concernées par l'expérimentation, à partir la banque de
données NCBI1 (National Center for Biotechnology
Information). Pour le choix des souches cibles, nous nous sommes fixés
dans un premier temps sur les souches pathogènes dont l'annotation a
été finie à savoir : Mt H37Rv, Mt CDC1551, Mt F11, Mt
H37Ra (voir Tableau 0.2). Par la suite les autres souches (voir
Tableau 0.3) peuvent êtres prises en compte au fur et à
mesure de leurs complètes annotations.
Ensuite, un prétraitement nécessaire est
effectué pour l'épuration, la préparation, et le formatage
des données afin de les rendre exploitables lors de
l'expérimentation. Les données brutes utilisées sont
constituées de plusieurs fichiers sous le format original i.e. texte
brut. Cette étape de prétraitement utilise le pseudo code suivant
:
Algorithme : Pretraitment_sequence
Début
Entrée : le fichier des gènes
(fichier_gene)
Sortie : le fichier des gènes nettoyé
(fichier_gene_nettoyé)
Variables : ligne, sous_chaine ,code_gene, nom_gene,
nom_souche, Crochet1, Crochet2 : chaîne
taille, pos1, pos2 : entier
Crochet1="[" , Crochet2="]"
Lire (fichier_gene)
Tant que (NFF fichier_gene )
faire
Si ( ligne[0] = chiffre )
alors
code_gene =trouver_code_gene(ligne_courante)
ecrire_fichier(fichier_gene_nettoyé
,code_gene
Sinon
sous_chaine=ligne(debut,5)
Si ( sous_chaine # "Other" et sous_chaine #
"Annot" et sous_chaine #"This") alors
pos1=trouver_ligne(ligne,Crochet1) pos2=trouver_ligne(ligne,Crochet2)
nom_gene=trouver_gene(ligne,pos1) nom_souche=trouver_souche(ligne,pos1,pos2)
ecrire_fichier(fichier_gene_nettoyé ,nom_gene)
ecrire_fichier(fichier_gene_nettoyé ,nom_souche)
Finsi
Finsi FinTant que Fermer_fichier(fichier_gene)
Fermer_fichier(fichier_gene_nettoyé)
Fin
1
http://www.ncbi.nlm.nih.gov/Database/
http://www.ncbi.nlm.nih.gov/genomes/genlist.cgi?taxid=2&type=0&name=Complete%20Bacteria

Chapitre IV : Conception et expérimentation du
système BIODM - 58 -
Les Figures 4.2, et 4.3 montrent (en
flèche) sur quelques morceaux de fichiers, les descripteurs les plus
importants dans la définition d'une séquence
biologique.

1: aac
aminoglycoside 2-N-acetyltransferase [Mycobacterium
Tuberculosis CDC1551]
Other Aliases: MT0275
Annotation: NC_002755.2 (314424..314969, complement)
GeneID: 923198
4257: tRNA-Arg-4
tRNA [Mycobacterim Tuberculosis CDC1551]
Annotation: NC_002755.1 (4209177..4209249,
complement)
GeneID: 922623
This record was discontinued.
Figure 4.2 : Morceaux de la séquence
génomique du Mt CDC1551.
1: AAK44224
chromosomal replication initiator protein DnaA
[Mycobacterium Tuberculosis CDC1551]
gi|13879042|gb|AAK44224.1|[13879042]
2: AAK44225
DNA polymerase III, beta subunit [Mycobacterium
Tuberculosis CDC1551] gi|13879043|gb|AAK44225.1|[13879043]
7880: NP_338144
virulence factor mce family protein [Mycobacterium
Tuberculosis CDC1551] gi|15843107|ref|NP_338144.1|[15843107]
Figure 4.3 : Morceaux de séquence
protéique du Mt CDC1551.
2ème étape : Transformation des
données
La transformation des données, du format
original vers un formalisme base de données (attribut, valeur), est
faite à partir des descripteurs possibles dégagés
auparavant. Cependant, les caractéristiques dégagées n'ont
pas des valeurs binaires, ce qui nécessitera une opération de
« binarisation » afin que les algorithmes d'extraction de
règles puissent les traiter. Il y a évidemment de très
nombreuses manières d'effectuer cette "binarisation", celle
adoptée par notre processus est de vérifier la présence ou
non du gène ou de la protéine dans les séquences en
question. Ainsi, un gène présent sera noté "1" et absent
par "0". On peut aussi proposer de choisir d'autres valeurs, néanmoins
nous pouvons dire que cette étape de prétraitement est longue,
complexe, et nécessite de
Chapitre IV : Conception et expérimentation du
système BIODM - 59 -
faire de nombreux choix.
De plus, il est difficile de déterminer dans
quelle mesure ces choix ont une influence sur le résultat des
extractions de connaissances. Les traitements de cette étape sont
décrits par le pseudo code suivant :
Algorithme : Transformation
Début
Entrée : fichier des gènes nettoyé
(fichier_gene_nettoyé)
Sortie : table des gènes (T_gene)
chaine ligne_courante
Lire (fichier_gene_nettoyé)
Tant que (NFF fichier_gene_nettoyé)
faire
Cas de ( lig )
lig =1 : xcode=ligne_courante , lig=2
lig =2 : xgene=ligne_courante , lig=3
lig =3 : xsouche=ligne_courante , lig=1
Fin cas
ecrire_table(T_gene, xcode,xgene, xsouche)
Fin Tant que
Fermer_fichier(fichier_gene_nettoyé)
Fermer_table(T_gene)
Fin
3ème étape : Production et
évaluation des règles d'association
Production. Cette phase concerne l'utilisation de
l'algorithme d'extraction de motifs sur les données
préparées pendant l'étape précédente. Dans
cette phase, l'utilisateur doit définir les contraintes sur ces motifs
et fixer les paramètres de l'algorithme i.e. support et confiance. Cette
définition des contraintes est importante car souvent l'ensemble des
motifs possibles est tellement grand qu'il n'est pas calculable, il faut donc
le restreindre.
L'algorithme Apriori est alors utilisé pour
rechercher les Itemsets. Ensuite un traitement approprié est
effectué sur les Itemsets valables afin de trouver les règles
d'association par une opération de combinaison.
Evaluation. La production des règles
d'association peut donner des règles non intéressantes, il est
donc nécessaire d'évaluer leurs qualités. Cette
évaluation sera faite en utilisant la confiance et le support pour ne
retenir que les règles dont le support et la confiance dépassent
les seuils fixés par l'utilisateur.
Il existe aussi d'autres méthodes
d'évaluation qui viennent renforcer le choix des règles
produites. Ces méthodes utilisent des fonctions d'évaluations,
elles ne sont pas abordées dans le cadre de cette
étude.
A un autre niveau, l'évaluation du
spécialiste est nécessaire. En effet, les algorithmes
Chapitre IV : Conception et expérimentation du
système BIODM - 60 -
d'extraction de motifs fréquents ou de
construction de modèles permettent de découvrir des
propriétés sur les données. Néanmoins, ces
propriétés ne peuvent être considérées comme
de nouvelles connaissances tant qu'elles n'ont pas été
interprétées et validées par un expert du domaine. Ces
propriétés découvertes par les algorithmes peuvent
être non intéressantes, incomplètes voir même
provenir d'erreurs dans les données, mais seul le spécialiste du
domaine saura les interpréter. Les traitements de cette étape
sont décrits par le pseudo code suivant :
Algorithme : Production des règles
d'association Début
Entrée : table des gènes
(T_gene)
Sortie : fichier des règles d'association
(fichier_RA)
fichier_RA = Apriori (T_gene)
Fermer_fichier(fichier_RA ) Fermer_table(T_gene)
Fin
4eme étape : Transformation des règles
d'association
Les règles trouvées sont
transformées par élagage de toutes les informations inutiles puis
représentées selon un formalisme transitoire aidant par la suite
à la production d'un graphe d'induction, nécessaire au passage
à une modélisation par le principe booléen. Une
règle d'association Ri se verra traduite en une règle
transitoire Rti selon le schéma suivant :

( Ri , Antécédenti , Conséquenti
, support , confiance)

Rti , { Prémissei (
Antécédenti ) } , { Conclusioni ( Conséquenti )
}
Les traitements de cette étape sont décrits
par le pseudo code suivant :
Algorithme : Transformation_RA
Début
Entrée : fichier des règles d'association (
fichier_RA )
Sortie : fichier des règles transitoires (
fichier_RT )
chaine ligne_courante
Lire (fichier_RA)
Tant que (NFF fichier_RA)
faire
Num_regle= extraire_num_regle(ligne_courante)
Prémisse= extraire_antecedent(ligne_courante) Conclusion=
extraire_conclusion(ligne_courante) ecrire_fichier(Fichier_RT, Num_regle,
Prémisse, Conclusion)
Fin Tant que
Fermer_fichier(fichier_RA) Fermer_fichier(fichier_RT)
Fin
Chapitre IV : Conception et expérimentation du
système BIODM - 61 -
5eme étape : Production du graphe
d'induction
Les règles transitoires vont aider à
construire un graphe d'induction par un traitement approprié selon le
principe suivant : un sommet désigne un noeud sur lequel on fait un
test, avec les résultats possibles binaires ou multivalués.
L'algorithme suivant en pseudo code illustre la production de ce graphe. Les
traitements de cette étape sont décrits par le pseudo code
suivant :
Algorithme : Création_Graphe
Début
Entrée : fichier des règles transitoire
(Fichier_RT)
Sortie : fichier du graphe (Fichier_Graphe)
Chaine sommet1, sommet2
Fichier intermédiaire ( Fichier_inter)
Lire (fichier_RT)
Tant que (NFF Fichier_RT)
faire
P=Extraire_ prémisse (ligne_courante)
C=Extraire_conclusion (ligne_courante)
ecrire_fichier (fichier_inter, P, " ", C, "
")
Fin tantque
Initialiser_graphe (Fichier_Graphe)
Lire (fichier_inter)
Tant que (NFF fichier_inter)
faire
Sommet1=Calculer_noeud_depart (P)
Sommet2=Calculer_noeud_arrivée (C)
ecrire_noeud_graphe (sommet1, sommet2)
Fin tantque
Effacer (Fichier_inter)
Fermer (Fichier_RT)
Fermer (Fichier_Graphe)
Fin
6eme étape : Production des règles
cellulaires
(1) Génération des règles
cellulaires
A partir du graphe d'induction, on produira les
règles cellulaires sous la forme :
Rck : Si { Prémissek } Alors { Conclusionk ,
Sommetk }
où Prémissek et Conclusionk
sont composées des Items de la règle d'association, et
Sommetk
le noeud du graphe d'où est produite la
règle cellulaire.
(2) Représentation cellulaire
Les règles générées
auparavant (étape 6.1) sont représentées en couches
cellulaires. Les Items des Prémissek , des Conclusionk
et les Sommetk vont constituer les faits (CELFAIT) et les Rck
vont constituer les règles (CELREGLE).
Schématiquement nous aurons :
{ Rck } CELREGLE et { Prémissek , Conclusionk,
, Sommetk } CELFAIT
Chapitre IV : Conception et expérimentation du
système BIODM - 62 -
avec k E [1, n]. Ces ensembles
seront représentés sous le format de couches cellulaires suivant
:
|
REGLES
|
|
|
|
|
|
|
ER
|
IR
|
SR
|
|
0
|
1
|
1
|
|
...
|
|
|
|
Les couches
|
CELFAIT
|
et CELREGLE
|
|
CELFAIT
|
FAITS
|
CELREGLE
|
|
EF
|
IF
|
SF
|
|
Fait1
|
1
|
0
|
0
|
R1
|
|
0
|
1
|
0
|
R2
|
|
Faitsn
|
|
|
|
|
Les traitements de cette étape sont décrits
par le pseudo code suivant :
Algorithme : Production des règles cellulaires
Début
Entrée : fichier graphe
(Fichier_Graphe)
Sortie : le fichier des faits (fichier_fait)
le fichier des regles (fichier_regles)
les fichiers CELFAIT, CELREGLE, RE, RS
Lire (Fichier_Graphe)
Pour chaque noeud
Sommet=noeud
Suc = extraire_Suc (noeud)
val = extraire_val (Suc)
ecrire_fait (fichier_fait , sommet, val, suc,
type)
ecrire_regle (fichier_regles, sommet_noeud, val,
Suc)
Fin pour
Pour chaque regle
Fait=Extraire_fait (regle)
type=Calculer_ type (Fait)
Si (type=valeur) Alors if = "1" Fin
si
Si (type=sommet) Alors if = "0" Fin
si
Si (fait="s0") Alors ef = "1" Sinon
ef= "0" Fin si
Ecrire_CELFAIT (Fait, if, ef, "0")
Num_regle=Extraire_num_regle (regle)
Ecrire_CELREGLE (Num_regle,
"0","1","1" )
Fin Pour
Pour chaque fait
type=Extraire_ type (fait)
num_regle=Extraire_num_regle (fait)
Si (type="Prémisse") Alors
Re = "1" sinon Re="0" Fin si
Ecrire_RE (fait, num_regle, Re)
Si (type="Conclusion") Alors
Rs = "1" sinon Rs="0" Fin si
Ecrire_RS (fait, num_regle, Rs)
Fin Pour
Fermer_fichier Fichier_Graphe, fichier_fait ,
fichier_regles , CELFAIT, CELREGLE, RE, RS
Fin
Chapitre IV : Conception et expérimentation du
système BIODM - 63 -
7eme étape :
Intégration
La machine cellulaire intégrera et exploitera
la représentation cellulaire et les matrices d'E/S à travers une
inférence en chaînage avant (recherche des règles
applicables, choix d'une règle à appliquer, ajouter la conclusion
à la base de faits pour enrichir la base de connaissances).
La dynamique de la machine cellulaire utilise les deux
fonctions de transition citées auparavant (Chapitre III - Section 1),
à savoir :
· La fonction de transition äfact
:
äfact (EF, IF, SF, ER, IR, SR) = (EF, IF, EF,
ER+(RET.EF), IR, SR)
· La fonction de transition ärule
:
ärule (EF, IF, SF, ER, IR, SR) = (EF+(
RS.ER), IF, SF, ER,
IR, ^ER)
Nous considérons G0 la configuration initiale
de notre machine cellulaire et que ?=ärule
° äfact la fonction de transition globale :
?(G0)=G1 si äfact(G0)=G'0 et
ärule(G'0)=G1. Supposons que G={G0, G1,
....,Gq} est l'ensemble des configurations de la machine cellulaire.
L'évolution discrète de la machine d'un génération
à une autre est définie par la séquence G0, G1,
....,Gq, oÙ Gi+1
=?(Gi).
IV.4 Le logiciel réalisé
IV.4.1 Présentation fonctionnelle du logiciel
Le logiciel que nous avons réalisé
permet de produire des règles d'association, ensuite celles-ci subiront
un post-traitement pour produire des règles booléennes
inductives, ceci bien entendu avec une visualisation des résultats. Le
schéma que nous présentons montre les fonctionnalités sans
pour autant fixer une quelconque chronologie dans les opérations. Ce
schéma représente les principales classes java et les
différentes interactions possibles entre elles, i.e. une classe
supérieure (schématiquement) interagit avec les classes
subordonnées à l'aide de fonctions ou procédures internes.
Les classes de moindres importances ne seront pas explicitées pour
présenter un schéma simplifié et
compréhensible.
Chapitre IV : Conception et expérimentation du
système BIODM - 64 -
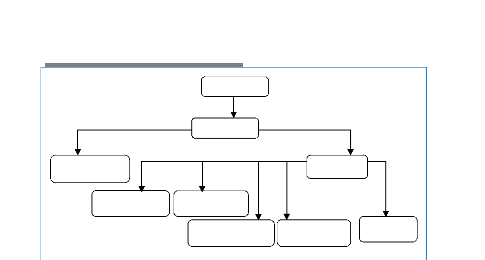
BIODM
Frame.One
Experiment
Explore_
Biological_Data
Association_
Rules_Procedure
Data_Base_ Procedures
Graph_Induction_ Creation
Cellular_Rules_ Generation
Save_
Experiment
Figure 4.4 : Architecture fonctionnelle du
système BIODM.
IV.4.2 Les différentes classes
(1) Classe BIODM
C'est la classe qui lance toute l'application. Elle
ne contient qu'une instance de la classe FRAME ONE.
(2) Classe FRAME_ONE
C'est la fenêtre principale du programme. Elle
contient trois panels (Selection_Panel, Panel1, Panel2) et une barre de menu.
Toutes les opérations passent par celle-ci. C'est la classe la plus
importante du système car elle gère toutes les opérations
sur les séquences biologiques par le biais de la barre de menu. Elle
contient trois Jtable avec scrollbars et permet l'affichage des
résultats de l'expérimentation.
(3) Classe EXPLORE_BIOLOGICAL_DATA
Elle permet de visualiser les données
expérimentales (séquences biologiques) pour une possible
vérification ou pour simple lecture des données avant
sélection de celles-ci par l'utilisateur.
(4) Classe EXPERIMENT
C'est la classe qui permet de démarrer
l'expérimentation à l'aide d'une boite de dialogue qui permet
à l'utilisateur de sélectionner les fichiers choisis pour
l'expérimentation.
Chapitre IV : Conception et expérimentation du
système BIODM - 65 -
(5) Classe ASSOCIATION_RULES _PROCEDURES
C'est la classe qui contient toutes les
méthodes de production des règles d'association telles que la
recherche des Itemsets, calcul de fréquence, calcul de support. Elle
permet la recherche des règles d'association. Elle fait appel à
ses propres méthodes et à celles stockées dans la classe
DATA_BASE_PROCEDURES. Cette classe fait aussi appel à une méthode
Visualisation_Regles, qui permet de présenter à l'utilisateur les
règles d'association obtenues, sous une forme textuelle. Une
possibilité de validation des règles est offerte à
l'utilisateur, pour décider de sauvegarder son expérimentation ou
pas.
(6) Classe DATA_BASE_PROCEDURES
Elle regroupe toutes les méthodes de gestion
de la base de données telles que la création d'une connexion
à la base de données, la fermeture de la connexion, lecture et
écriture dans une table, et toutes les différentes
requêtes.
(7) Classe GRAPHE_INDUCTION_CREATION
Elle permet de créer le graphe d'induction en
utilisant les règles d'association transformées pour créer
un graphe sur lequel se base la classe CELLULAR_RULES_GENERATION pour produire
des règles cellulaires.
(8) Classe CELLULAR_RULE_ GENERATION
Elle permet de créer les règles
cellulaires et de les intégrer dans la base de connaissances selon le
formalisme expliqué auparavant. Elle contient toutes les méthodes
de création des couches CELFAIT et CELREGLE.
(9) Classe SAVE_EXPERIMENT
Elle permet de sauvegarder les résultats de
l'expérimentation, sous les formats appropriés pour chaque type
de donnée à savoir le format texte et base de données.
L'utilisateur aura ainsi la possibilité de visualiser les
résultats de son expérimentation
ultérieurement.
IV.4.3 L'interface
L'interface permet à l'utilisateur d'explorer
les séquences de données biologiques manipulées ceci afin
de lire directement les données sur une séquence
choisie.
De plus, l'utilisateur a la possibilité
d'expérimenter l'ECD sur les séquences qu'il aura bien avant
sélectionnées, puis paramétrera son expérimentation
à travers le support
Chapitre IV : Conception et expérimentation du
système BIODM - 66 -
et la confiance. Une fois l'expérimentation
réalisée, les résultats sont affichés pour une
interprétation possible par l'expert du domaine.
Figure 4.5 : Interface du système
BIODM.
IV.5 L'expérimentation
Nous avons implémenté l'algorithme
Apriori en langage JAVA sur une plate-forme Windows. Les tests
expérimentaux ont été conduits sur une machine Intel
Celeron CPU 540 à 186 GHz 512 Mo RAM. Pour examiner l'efficacité
pratique de notre algorithme, nous avons mené une sérié
d'expérimentations sur des données réelles, dont les
caractéristiques sont détaillées dans ce qui
suit.
La base de test
En se basant sur les souches complètement
annotées (voir Tableau 0.2), que nous avons
considérées comme souches échantillon, nous relevons le
nombre de gènes total par souches ainsi que les gènes constituant
l'échantillon de l'ECD sur le Tableau 4.1 et la Figure
4.6.
|
Souche
|
Nombre de
gènes existants
|
Nombre de gènes de
l'échantillon
expérimental
|
|
1
|
Mt CDC1551
|
4293
|
20
|
|
2
|
Mt F11
|
3998
|
20
|
|
3
|
Mt H37Ra
|
4084
|
20
|
|
4
|
Mt H37Rv
|
4048
|
20
|
Tableau 4.1 : Base de test servant à
l'expérimentation.
Chapitre IV : Conception et expérimentation du
système BIODM - 67 -

Nous nous sommes fixés un nombre convenable et
suffisant pour notre expérience vu le problème combinatoire de
l'algorithme qui génère un nombre assez important d'Itemsets
fréquents à chaque itération. Nous avons alors pris les 15
premiers de chaque séquence, avec la supposition que ces quinze
gènes sont assez représentatifs et distinctifs de chaque souche
prise séparément. Par la suite l'expérience pourra
être tentée en réel sur une machine suffisamment puissante
du genre station de travail.
1 ) aac accD aceA-1 aceA-2 aceB aceE ackA acnA acp-1
acp-2 acpP acpS acs adh adk
2 ) aceE acpP acpS adk alaS alr argC argD argJ argS aroB
aroE aroK aspS atpC
3 ) aac aao accA1 accA2 accA3 accD1 accD2 accD3 accD4
accD5 accD6 aceAa aceAb aceE acg
4 ) 35kd_a aac aao accA1 accA2 accA3 accD1 accD2 accD3
accD4 accD5 accD6 aceAa aceAb aceE
Figure 4.6 : Echantillon de gènes servant
à la fouille de données.
Les résultats expérimentaux
On se basant sur l'échantillon
expérimental cité auparavant (voir Figure 4.6), le
système BIODM donne des résultats intéressants qui
resteront à consolider avec de nouvelles souches en cours de
séquençage (voir Tableau 1.3) et qui seront prises en
considération par notre système au fur et mesure de leur
publication définitive sur leurs sites d'origines, NCBI.
De plus, d'après l'algorithme Apriori, un
principe de base de la génération de règles est celui de
la génération de combinaisons possibles d'Items pour trouver les
Itemsets, ce traitement prend énormément de temps, ce qui risque
de mobiliser la machine pour un temps assez conséquent, du fait
qu'à chaque itération il génère 2n
Itemsets possibles.
Ainsi, par exemple pour 15 gènes nous notons un
ordre de grandeur de plus de 835000 de règles avec un temps
d'exécution machine de avoisinant les 5 secondes, ce qui est
énorme. Ce qui à fait que nous avons limité notre
échantillon d'apprentissage à une vingtaine de gènes i.e.
Items, ceci pour éviter un temps de calcul énorme pour une
machine modeste. Nous avons aussi noté la constatation suivante
:
Plus l'échantillonnage est grand (> 15),
plus le temps de calcul croit énormément arrivant jusqu'à
réduire le temps de réponse de notre machine.
Chapitre IV : Conception et expérimentation du
système BIODM - 68 -
|
Prémisse -> Conclusion
|
Support
|
Confiance
|
|
acpP -> aceE
|
100.0
|
100.0
|
|
aac ackA - > aceE
|
75.0
|
100.0
|
|
aac aceE -> ackA
|
75.0
|
100.0
|
|
ackA aceE -> aac
|
75.0
|
100.0
|
|
aac ackA -> acpP
|
75.0
|
100.0
|
|
aac acpP -> ackA
|
75.0
|
100.0
|
|
ackA acpP -> aac
|
75.0
|
100.0
|
|
aac aceE -> acpP
|
75.0
|
100.0
|
|
aac acpP -> aceE
|
75.0
|
100.0
|
|
ackA aceE -> acpP
|
75.0
|
100.0
|
|
ackA acpP -> aceE
|
75.0
|
100.0
|
|
acpS aceE -> acpP
|
75.0
|
100.0
|
|
acpS acpP -> aceE
|
75.0
|
100.0
|
|
aac ackA aceE -> acpP
|
75.0
|
100.0
|
|
aac ackA acpP -> aceE
|
75.0
|
100.0
|
Tableau 4.2 : Exemple de règles
générées par Apriori pour un support
de 60% et une
confiance de 80%.
|
Règles cellulaires
|
|
Rc1 : s0 -> aac=1 , s1
|
|
Rc2 : s0 -> accA1=1 , s2
|
|
Rc3 : s0 -> accA2=1 , s3
|
|
Rc4 : s0 -> accA3=1 , s4
|
|
Rc5 : s0 -> accD1=1 , s5
|
|
Rc6 : s0 -> accD2=1 , s6
|
|
Rc7 : s0 -> accD3=1 , s7
|
|
Rc8 : s0 -> accD4=1 , s8
|
|
Rc9 : s0 -> accD5=1 , s9
|
|
Rc10 : s0 -> accD6=1 , s10
|
|
Rc11 : s11 -> aac=1 , s12
|
|
Rc12 : s11 -> accA2=1 , s13
|
Tableau 4.3 : Exemple de règles cellulaires
générées par BRI.
Chapitre IV : Conception et expérimentation du
système BIODM - 69 -
Temps de traitement.
En utilisant la base de test, le système BIODM
donne des résultats intéressants et montre l'évolution
exponentielle du temps d'exécution (voir tableau 4.4). Nous
pouvons constater également que l'exécution de l'algorithme
Apriori prend une part importante en temps d'exécution du système
en totalité, i.e. dans ses phases les plus importantes de
l'expérimentation à savoir : la génération des
règles d'association par Apriori et la génération des
règles booléennes par BRI.
|
Confiance %
|
Support %
|
Nombre de Gènes
|
Items générés
|
Nombre de règles
|
Temps d'exécution Apriori
|
Temps d'exécution global
|
|
10
|
70
|
15
|
41
|
69
|
0.00 s
|
0.00 s
|
|
30
|
50
|
15
|
41
|
147701
|
0.86 s
|
2.23 s
|
|
50
|
30
|
15
|
41
|
147687
|
0.86 s
|
2.23 s
|
|
70
|
10
|
15
|
41
|
835284
|
4.92 s
|
10.26 s
|
Tableau 4.4 : Nombre de règles et temps
d'exécution d'Apriori sur l'échantillon Figure
4.6.
Espace de stockage.
Nous constatons qu'à titre indicatif pour un
ensemble de 147687 règles d'association, le fichier occupe un espace de
stockage de 8.65 MO alors que pour les règles cellulaires
correspondantes, le fichier est de 6.13 MO.
Nous constatons qu'une représentation
cellulaire est plus intéressante du point de vue espace de stockage et
que sur un ensemble de règles encore plus conséquent, nous aurons
un gain en espace de stockage assez significatif qui se répercutera
positivement sur la performance du système, et mentionnons là
aussi que ce ne sont que des résultats partiels qu'il faudra consolider
avec un échantillon plus important.
|
Nombre de règles
d'association
produites
|
Espace de Stockage
(règles
d'association)
|
Espace de stockage
(représentation
booléenne)
|
|
69
|
1.78 KO
|
0. 81 KO
|
|
147701
|
8.65 MO
|
6.11 MO
|
|
147687
|
8.65 MO
|
6.12 MO
|
|
835284
|
48.8 MO
|
39.14 MO
|
Tableau 4.5 : Evolution de l'espace de
stockage.
Chapitre IV : Conception et expérimentation du
système BIODM - 70 -
IV.6 Conclusion
Nous avons défini un système qui permet
à un spécialiste du domaine, en l'occurrence le biologiste, de
procéder à la fouille de données dans un contexte
biologique. Nous avons pu procéder à des tests selon une
configuration machine bien modeste, néanmoins nous signalons qu'un
problème de taille auquel nous étions confronté celui des
données biologiques ciblés qui ne sont pas toujours disponibles
en continue vu le problème d'annotation des génomes qui prend du
temps. C'est pour cela que nous avons bien choisi nos échantillons de
test et nous avons pu vérifier, combien même avec un
échantillonnage réduit, le processus est couteux en temps de
traitement.
Enfin, nous pouvons dire que le système
prototype que nous venons de réaliser était dans un objectif
expérimental. Nous avons proposé une interface simple qui montre
les principales manipulations que peut faire un biologiste pour procéder
aisément à une fouille de données biologiques.
Conclusion générale - 71 -
Conclusion générale
Nous avons abordé dans ce mémoire une nouvelle
problématique qui consiste à fouiller des données
biologiques avec post-traitement des règles extraites pour optimiser le
stockage et la gestion. L'objectif que nous nous sommes fixé
était de proposer une solution appuyée par des travaux
antérieurs de modélisation booléenne selon le principe
cellulaire de la machine CASI.
Nous nous sommes basés sur une fouille à l'aide
de l'algorithme Apriori qui fonctionne par itération, i.e., l'extraction
des k-itemsets fréquents à partir des k-1 Itemsets
fréquents. Alors, n itérations sont faites pour extraire la
totalité des itemsets fréquents, avec n-1 la taille du plus long
itemset fréquent. A chaque itération, un ensemble d'items
candidats est généré à partir des itemsets
fréquents du niveau précédent.
Ceci dit, l'objectif de ce mémoire était aussi
de présenter l'état de l'art de notre thème de recherche,
de faire une étude comparative afin de soulever la problématique
de la représentation des données biologiques qui ne sont en fait
dans une structure de donnée standard, connue et théorisée
en informatique. Nous avons alors consacré le premier chapitre pour
l'ECD biologiques qui travaille sur un nouveau type de donnés si l'on
peut dire « non usuel » et nous avons visé
particulièrement des données se rapportant à une
épidémie qui est la tuberculose afin de tester
ultérieurement en réel, ce qui nous à fait faire une
prospection de ces données et tirer des constatations utiles pour la
suite de notre étude surtout pour les étapes nettoyage et
prétraitement de l'ECD.
Ensuite, nous avons fait une étude comparative et avons
abordé l'extraction des règles d'association, et leur
optimisation par le module BRI qui constitue le support de notre solution pour
le post-traitement en fouille de données.
Notre étude se voulait être assez novatrice, dans
la mesure où nous avons été incités à
utiliser des techniques prouvées de la machine cellulaire CASI,
combinées à une fouille de données. De ce fait, deux
objectifs nous ont guidés dans la proposition d'un automate cellulaire
pour l'optimisation, la génération, la représentation et
la gestion d'une base de règles d'association. En effet, le premier
c'est d'avoir une base de règles optimisée et des temps de
traitements assez réduits grâce aux principes de
représentation booléenne, et le deuxième c'est d'apporter
une contribution à la construction des systèmes à base de
connaissances en adoptant une nouvelle technique
Conclusion générale - 72 -
cellulaire.
Perspectives
La solution que nous avons préconisée utilise
Apriori couplé avec un post-traitement des règles d'association
par une modélisation booléenne. La perspective que nous nous
proposons est de tester le système avec d'autres algorithmes, tels
qu'AIS et FP-Growth, afin de comparer les résultats du point de vue gain
de temps. Ensuite on pourra aussi faire guider le processus de fouille par le
spécialiste du domaine en introduisant par exemple ces
préférence par un ou plusieurs paramètre qui seront pris
en compte lors du processus de fouille, ceci afin de limiter la taille des
résultats qui comme nous le savons est très volumineux.
L'idée est de faire un processus d'ECD biologiques sous contrainte. Ce
volet nous semble prometteur vu les travaux de recherches déjà
faits avec ce concept.
Et finalement, nous pensons carrément à adapter
la modélisation booléenne à l'algorithme Apriori, et
proposer un automate cellulaire pour la fouille de données.
Références bibliographiques - 73
-
Références bibliographiques
[Abdelouhab et atmani., 2008]
Abdelouhab, F. _ Atmani, B.: Intégration
automatique des données semi-structurées dans un entrepôt
cellulaire, Troisième atelier sur les systèmes
décisionnels, 10 et 11 octobre 2008, Mohammadia - Maroc, pp. 109-120.
(2008).
[Agrawal et al., 1993]
Agrawal, R. _ Swami, A. _ Imielinski, T.: Mining
Association Rules Between Sets of Items in Large Databases. In Proceedings ACM
SIGMOD International Conference on Management of Data, pp207, Washington DC
(1993).
[Agrawal et Srikant., 1994]
Agrawal, R. _ Srikant, R.: Fast Algorithms for Mining
Association Rules. In Proceedings of the 20th International Conference on Very
Large Data Bases (VLDB'94), pp 487-499, Santiago, Chile (1994).
[Atmani et Beldjilali, 2007]
Atmani, B. _ Beldjilali, B.: Knowledge Discovery in
Database : Induction Graph and Cellular Automaton. Computing and Informatics
Journal, Vol. 26 N°2 171-197. (2007).
[Bahar et Chen, 2004]
Bahar, I. _ Chen, S.S.: Mining frequent patterns in
protein structure: a study of protease family. Vol. 20 Suppl. 1, pages i1-i9
DOI: 10.1093/bioinformatics/bth912 (2004).
[Benabdeslem et al., 2007]
Benabdeslem, K. _ Lebbah, M. _ Aussem, A. _ Corbex.
M.: Approche connexionniste pour l'extraction de profils cas-témoins du
cancer du Nasopharynx à partir des données issues d'une
étude épidémiologique", EGC 2007, RNTI, 2007 Volume E-9
N°2, Pages : 445-454 (2007).
[Benamina et Atmani, 2008]
Benamina, B. _ Atmani, B.: WCSS: un système
cellulaire d'extraction et de gestion des connaissances, Troisième
atelier sur les systèmes décisionnels, 10 et 11 octobre 2008,
Mohammadia - Maroc, pp. 223234. (2008).
[Boutin et al., 2003]
Boutin, J.P. _ Spiegel, A. _ Michel, R. _ Ollivier, L.:
Les mesures d'association en épidémiologie. Med Trop; 63 : 75-78
(2003).
[Broad]
Broad Institute.:
http://www.broad.mit.edu
[Callas, 2009]
Calas, G.: Études des principaux algorithmes de
data mining. SCIA EPITA (2009).
[Carbonelle et al., 2003]
Carbonnelle, B. _ Dailloux, M. _ Lebrun, L. _ Maugein, J.
_ Pernot, C.: Cahier de formation en biologie médicale N°29
(2003).
[Chen et al., 2003]
Chen, H. _ Fuller, S.S. _ Friedman, C. _ Hersh, W.:
Knowledge management, data mining, and text mining in medical informatics.
Medical Informatics, volume 8, Springer US. (2003).
Références bibliographiques - 74
-
[Chervitz et al., 1999]
Chervitz, S.A. _ Hester, E.T., Ball, C. _ Dolinski, K.
_ Dwight, S.S. _ Haris, M.A. _ Juvik, G. _ Malekian, A. _ Roberts, S. _ Roe T.
_ Cafe, C., Shroeder, M. _ Sherlock, G. _ Weng, S. _ Zhu, Y. _ Cherry, J.M. _
Botstein, D.: Using the Sacharomyces genome databases (SGD) for analysis of
protein similarities and structure ( Nucleic Acids Research, Vol 27 N° 1).
(1999).
[Denison et al., 1998]
Denison, D.G. _ Mallick, B.K _ Smith. A. F. M.: A
Bayesian CART algorithm. Biometrika, 85:363 377, 1998.
[Elloumi et Maddouri, 2002]
Elloumi, M. _ Maddouri, M.: A Data Mining Approach
based on Machine Learning Techniques to Classify Biological Sequences,
Knowledge Based Systems Journal, Vol. 15, Issue 4, Elsevier Publishing Co.,
Amsterdam, North-Holland (Publisher) : p217-223,(Mai 2002).
[Etienne, 2004]
Etienne, M.P.: Une approche statistique pour la
détection de gènes impliqués dans les maladies
multifactorielles. Actes de Jobim juin (2004).
[Fayyad et al., 1996]
Fayyad, U. _ Piatetsky-Shapiro, G. _ Smyth, P.: From
Data Mining to Knowledge Discovery: An Overview. In Fayyad, U. _ Piatetsky-
Shapiro, G. _ Amith, Smyth, P. _ and Uthurnsamy, R.: (eds.) Advances in
Knowledge Discovery and Data Mining. MIT Press, 1-36, Cambridge
(1996).
[Ferdinand et al., 2004]
Ferdinand, S. _ Valetudi, G. _ Sola, C. _ Rastogi, N.
: Data mining of mycobacterium Tuberculosis complexe genotyping results using
mycobacterial intersepted repetitive units validates the clonal structure of
spolygotyping-defined families. Research in Microbiology 155(8): 647-654
(2004).
[Fleiishman et al, 2002]
Fleiishman, R.D. _ Alland, D. _ Eisen, J.A _
Carpenter, L. _ White, O. _ Petersen, J. _ Deboy, R. _ Dodson, R. _ Gwinn M. _
Haft, D. _ Hickey, E. _ Kolonay, J.F. _ Nelson, W.C. _ Umayam, L.A. _
Ermolayeva, M. _ Salzberg, S.L. _ Delcher, A. _ Utterback, T. _ Weidman, J. _
Khouri, H. _ Gill, J. _ Mikula, A. _ Bishai, W. _ Jacobs, W.R. _ Venter, J.C. _
Fraser, C.M.: Whole-Genome comparaison of Mycobacterium Tuberculosis clinical
and laboratory stains. Journal of Bacteriology, October 2002, p. 5479-5490,
Vol. 184, No. 19 (2002).
[Gaussier, 2009]
Gaussier, E. : Arbres de décision Notes de cours
Université de Grenoble 1 - Lab. Informatique Grenbole
[Gibas et Jambeck, 2002]
Gibas, C. _ Jambeck, P.: Introduction à la
bioinformatique. Oreilly, ISBN10 : 2-84177-144-X (2002).
[Goebel, et Gruenland, 1999]
Goebel, M. _ Gruenwald. L.: A survey of Data Mining
and Knowledge Discovery Software Tools. ACM SIGKDD, Volume 1, Issue 1, page
20-33 (1999).
[Goethals et Van den Bussche, 2002]
Goethals, B. _ Van den Bussche, J.: Relational
Association Rules: Getting WARMeR. Proceedings of the ESFExploratory Workshop
on Pattern Detection and Discovery, pages 125-139, (2002).
[Han et Kamber, 1998]
Han, J. _ Kamber, M. : Data mining : Concepts and
techniques. SIGMOD'98, Seattle, Washington. (1998).
Références bibliographiques - 75
-
[Han et al., 2000]
Han, J., Pei, J., Yin, Y. « Mining Frequent Patterns
without Candidate Generation ». In Proceedings of
the 2000 ACM-SIGMOD Int'l Conf. On Management of Data,
Dallas, Texas, USA, May (2000).
[Hergalant et al., 2002]
Hergalant, S. _ Aigle, B. _ Leblond, P. _ Mari, J.F. _
Decaris, B.: Fouille de données à l'aide de HMM : application
à la détection de réitérations
intragénomiques. JOBIM'02, p.269-273, (2002).
[Hergalant et al., 2005]
Hergalant, S. _ Aigle, B. _ Leblond, P. _ Mari, J.F.:
Fouille de données du génome à l'aide de modèles de
Markov cachées. EGC 2005, Paris, France, Atelier fouille de
données complexes dans un processus d'extraction de connaissances, Jan
2005, p. 141 - 148. (2005).
[Hip et al., 2000]
Hipp, J. _ Güntzer, U. _ Nakhaeizadeh, G.:
Algorithms for Association Rule Mining - A General Survey and Comparaison. In
Proceedings, ACM SIGKDD 2000, Volume2, Issue 1, pp58-64 (2000).
[Labbe, 2007]
Labbe, A. : Introduction à
l'épidémiologie génétique. Notes de cours,
STT-66943. (2007).
[Larose, 2005]
Larose, D. T.: Discovering Knowledge in Data: An
Introduction to Data Mining. ISBN 0-471-666572 Copyright C_ 2005 John Wiley
& Sons, Inc. (2005).
[Maumus et al., 2005]
Maumus, S. _ A. Napoli, A. _ Szathmary, L.,
Visvikis-Siest. S.: Exploitation des données de la cohorte STANISLAS par
des techniques de fouille de données numériques et symboliques
utilisées seules ou en combinaison, in: Atelier Fouille de
Données Complexes dans un Processus d'Extraction des Connaissances - EGC
2005, Paris, France, Feb 2005, p. 73-76. (2005).
[Mhamdi et al., 2006]
Mhamdi, F. _ Elloumi, M. _ Rakotomalala, R.:
Extraction et Sélection des n-grammes pour le Classement de
Protéines, in Atelier Extraction et Gestion des Connaissances
Appliquées aux Données Biologiques, EGC-2006, Lille, pp.25-37,
Janvier (2006).
[Ncbi]
National Center for Biotechnolgy Information.:
http://www.ncbi.nlm.nih.gov.
[Oms]
Organisation Mondiale de la Santé.:
http://www.who.int/fr/
[Preux, 2008]
Preux, Ph.: Fouille de données : Notes de cours.
Université de Lille 3. (2008).
[Prum et Turi-Majoube, 2001]
Prum, B. _ Turi-Majoube, F.: Une approche statistique de
l'analyse des génomes. SMF - Gazette - 89, Juillet (2001).
[Quinlan, 1986]
Quinlan, J.R.: Induction on decision trees. Machine
Learning , vol. 1, pp. 81-106 (1986).
[Quinlan, 1993]
Quinlan, J.R. : C4.5: Programs for Machine Learning.
Morgan Kaufmann, San Mateo, CA, 1993.
Références bibliographiques - 76
-
[Rakotomalal, 1997]
Rakotomalala, R.: Graphes d'induction. Thèse de
DOCTORAT. Université Claude. Bernard - Lyon I. Décembre
(1997).
[Remvikos, 2004]
Remvikos, Y.: Epidémiologie analytique : Etudes
de cohortes. Med Trop pp 207-212. (2004).
[Sanger]
Sanger Institut.:
http://www.sanger.ac.uk.
[Savasere et al., 1995]
Savasere, A. _ Omiecinski, E. _ Navathe, S.: An
Efficient Algorithm for Mining Association Rules in Large Databases ». In
Proceedings of the 21th conference on VLDB (VLDB'95), Zurich, Switzerland
(1995).
[Tzanis et al., 2005]
Tzanis, G. _ Berberidie, C. _ Vlahavas, I.: Biological
Data Mining. Encyclopedia of Database Technologies and Applications : 35-41.
(2005).
[Wikipedia]
Wikipedia,
http://www.fr.wikipedia.org.
[Wilkinson, 1992]
Wilkinson, L.: Tree Structured Data Analysis: AID,
CHAID and CART. Sun Valley, ID, Sawtooth/SYSTAT Joint Software Conference
(1992)
[Yokoyama et al., 2007]
Yokoyama, E. _ Kishida, K. _ Ishinohe, S.: Improved
Molecular Epidemiological Analysis of Mycobacteriem Tuberculosis Strains Using
Multi-Locus Variable Number of Tandem Repeats typing. Jpn. J. Infect. 60.
(2007).
[Zaki et al., 2004]
Zaki, M.J _ Shinichi, M. _ Rigoutsos, I.: Workshop on
data mining in bioinformatics. Report BioKDD04, SIGKDD Explorations. Volume
6,Issue 2 - Page 153-154 (2004).
[Zaki et al., 1997]
Zaki, M.J. _ Parthasarathy, S. _ Ogihara, M. _ Li, W.
: New Algorithms for fast discovery of Association Rules. In Proceedings of the
3rd Int'l Conference on KDD and data mining (KDD'97), Newport Beach,
California, (1997).
[Zuker, 2008]
Zucker, J.D.: Introduction à la fouille de
données en bioinformatique. Cours master EID- P13. IRD UR GEODES.
(2008).
Annexe B - 77 -
|
Souche du Mycobacterium Tuberculosis
|
|
Genome
sequencing
status
|
1: Mycobacteriem Tuberculosis '98-R604 INH-RIF-EM'
[Broad Institute] Strain for comparative analysis
|
|
draft assembly
|
2: Mycobacterium tuberculosis 02_1987 [Broad
Institute] Strain being sequenced for comparative analysis
|
|
draft assembly
|
3: Mycobacteriem Tuberculosis 210 [TIGR] Causative
agent of tuberculosis
Size: 4 Mb; Chromosome: 1
|
|
in progress
|
4: Mycobacteriem Tuberculosis 94_M4241A [Broad
Institute] Isolate from China
|
|
draft assembly
|
5: Mycobacteriem Tuberculosis C [Broad Institute]
Drug-susceptible strain
|
|
draft assembly
|
6: Mycobacteriem Tuberculosis CDC1551
[TIGR]
Causative agent of tuberculosis. Size: 4 Mb;
Chromosome: 1
|
|
complete
|
7: Mycobacteriem Tuberculosis EAS054 [Broad
Institute] Sequenced for comparative analysis
|
|
draft assembly
|
8: Mycobacteriem Tuberculosis F11 [Broad
Institute]
Predominant strain in South African epidemic Size: 4
Mb; Chromosome: 1
|
|
complete
|
9: Mycobacteriem Tuberculosis GM 1503 [Broad
Institute] Strain used for comparative genome analysis.
|
|
draft assembly
|
10: Mycobacteriem Tuberculosis H37Ra [Beijing
Genomics Institute] An attenuated strain used in mycobacterial virulence
research
|
|
draft assembly
|
11: Mycobacteriem Tuberculosis H37Ra [Chinese
National HGC, Shanghai/Fudan University, P.R. China, Shanghai/Johns Hopkins
University, Department of Molecular Microbiology & Immunology, Bloomberg
School of Public Health, USA, Baltimore]
An avirulent strain derived from its virulent parent
strain H37 Size: 4 Mb; Chromosome: 1
|
complete
|
12: Mycobacteriem Tuberculosis H37Rv [Sanger
Institute]
Causative agent of tuberculosis. Size: 4 Mb;
Chromosome: 1
|
|
complete
|
13: Mycobacteriem Tuberculosis KZN 1435 [Broad
Institute] Multidrug-resistant clinical isolate
|
|
draft assembly
|
14: Mycobacteriem Tuberculosis KZN 4207 [Broad
Institute] Drug-susceptible clinical isolate
|
|
draft assembly
|
15: Mycobacteriem Tuberculosis KZN 605 [Broad
Institute] Extensively drug-resistant clinical isolate
|
|
draft assembly
|
16: Mycobacteriem Tuberculosis T17 [Broad
Institute] Strain will be sequenced for comparative genome
analysis
|
|
draft assembly
|
17: Mycobacteriem Tuberculosis T85 [Broad
Institute] Susceptible strain
|
|
draft assembly
|
18: Mycobacteriem Tuberculosis T92 [Broad
Institute] Clinical isolate
|
|
draft assembly
|
19: Mycobacteriem Tuberculosis str. Haarlem [Broad
Institute] A drug resistant strain found in crowded human
populations
|
|
draft assembly
|
|
Tableau 0.1 : Les différentes souches du
Mycobacterium Tuberculosis. [Source NCBI]1.
(*) Draft assembly = Projet(Contingent) d'assemblage
1
http://www.ncbi.nlm.nih.gov/Database/
http://www.ncbi.nlm.nih.gov/genomes/genlist.cgi?taxid=2&type=0&name=Complete%20Bacteria
Annexe B - 78 -
|
Souche
|
Taille(nt)2
|
protéines
|
gènes
|
Date création
|
Date maj
|
|
Mt CDC1551
|
4403837
|
4189
|
4293
|
Oct 2 2001
|
Jul 18 2008
|
|
Mt F11
|
4424435
|
3941
|
3998
|
Jun 14 2007
|
Jul 25 2008
|
|
Mt H37Ra
|
4419977
|
4034
|
4084
|
Jun 6 2007
|
Jul 9 2008
|
|
Mt H37Rv
|
4411532
|
3989
|
4048
|
Sep 7 2001
|
Jul 18 2008
|
Tableau 0.2 : Tableaux informatif sur ls
caractéristiques des souches du Mycobacterium
Tuberculosis
complètement annotées. [Source
NCBI]3.
|
|
Souches en cours d'annotation
|
|
·
|
Mt 10403-1
|
|
·
|
Mt 6404-1B
|
|
·
|
Mt 10403-10
|
|
·
|
Mt 6404-3B
|
|
·
|
Mt 10403-11
|
|
·
|
Mt 6404-A1
|
|
·
|
Mt 10403-4
|
|
·
|
Mt 7404-1
|
|
·
|
Mt 10403-7
|
|
·
|
Mt 7604-2
|
|
·
|
Mt 10403-8
|
|
·
|
Mt 7604-4
|
|
·
|
Mt 11105-2
|
|
·
|
Mt 7904-1
|
|
·
|
Mt 11105-3
|
|
·
|
Mt 7904-2
|
|
·
|
Mt 15304-1B
|
|
·
|
Mt 8104-1C
|
|
·
|
Mt 15304-3A
|
|
·
|
Mt 8104-2A
|
|
·
|
Mt 210
|
|
·
|
Mt subsp. tuberculosis
|
Tableau 1.3 : Les souches du Mycobacterium
Tuberculosis en cours d'annotation.
[Source NCBI]
2 nt :nucléotide.
3
http://www.ncbi.nlm.nih.gov


