Chapitre II
Matériel
et
Méthodes
1. Chapitre II Matériel et méthodes

15
Matériel végétal
Notre étude a été réalisée
sur une plante spontanée: le ricin (Ricinus communis). Les
échantillons sont collectés à partir de trois sites
nommés respectivement: Contrôle (C), le site 1 (S1: situé
à 100 m de l'origine de la pollution) et le site 2 (S2: situé
juste à côté de la source de la pollution : cimenterie de
Bir Mecherga).

Photo de la cimenterie à partir de Google
maps
2. Extraction et dosage des chlorophylles et des
caroténoïdes
Les teneurs en chlorophylles a, b et totales et en
caroténoïdes (mg.g-1 MF) ont été
déterminées selon la méthode de Lichtenthaler
(1988). Deux feuilles d'environ 100 mg de masse fraîche sont
pesées et mises dans 5 ml d'acétone (80 %). Après un
séjour de 72 heures à l'obscurité et à une
température de 4°C, la densité optique de l'extrait est
mesurée á 470 nm, 646 nm et 663 nm. Les teneurs en chlorophylles
a, b et totale et en caroténoïdes sont ensuite calculées
selon les équations suivantes:
Chlorophylle a = 12,25A663 - 2,79A646 Chlorophylle b =
21,50A646 - 5,10A663
Chlorophylle totale) = 7,15A663 + 18,71A646
Chapitre II Matériel et méthodes

16
Caroténoïdes = (1000A470 - 1,82Ca - 85,02Cb)
/198
3. Extraction et dosage du lycopène
Le lycopène est un pigment liposoluble, du fait de sa
grande disponibilité, il est beaucoup utilisé comme colorant
(E160d), c'est un tétra terpène de la famille des
carotènes. L'identification du lycopène se fait par
spectrophotomètre à 472 nm (Benakmoum et
al., 2008).
Une quantité de 100 mg de matière fraîche
est mise dans 10 ml d'un mélange formé de
hexane-acétone-éthanol (50/50/1). Le mélange est
agité pendant 10 min puis centrifugé à 5000 tours pendant
15 mn. Un volume de 1 ml de la phase organique est ensuite dilué dans 10
ml d'hexane. L'absorbance est mesurée à 472 nm.
4. Dosage du malondialdéhyde (MDA)
Le MDA est déterminé selon la méthode
décrite par Heath et Packer, (1968). Des
échantillons de 0,2 g sont broyés dans de l'azote liquide dans un
mortier en porcelaine. La poudre obtenue est homogénéisée
dans un mélange contenant 0.5% d'acide thiobarbiturique (TBA) et 20%
d'acide trichloroacétique (TCA). L'extrait est incubé à
95°C, pendant 30 min. Les tubes sont ensuite mis dans la glace puis
centrifugés à 4000 g, pendant 30 min à 4°C.
L'absorbance est mesurée à 532 et 600 nm. La concentration en MDA
(?mol g-1 MF) est calculée en utilisant le coefficient
d'extinction à 532 nm (155 mM cm-1).
5. Extraction et dosage des
mélanoïdines
Une quantité de 100 mg de tissu est extraite par
l'ajout de 1 ml d'eau distillée. La solution est placée dans un
rotateur, pendant 24h à 4°C puis centrifugée à 1300 g
pendant 15 min. Le surnageant est transféré dans de nouveaux
tubes. L'extrait est scanné avec un UV spectrophotomètre, selon
un spectre de 200 à 700 nm. Le pic est obtenu à 360 nm, comme a
été décrit par Shin et al.,
(2009).
6. Activité antioxydante
6.1. Extraction des protéines solubles
Après broyage des échantillons (1g) dans l'azote
liquide, la poudre est reprise dans un tampon d'extraction (1,5 ml) dont son
volume est proportionnel à la masse de matière fraîche et
dont la composition est la suivante:

17
|
Chapitre II
|
Matériel et méthodes
|
|
Phosphate de potassium
|
50 mM, pH 7.5
|
|
PVPP
|
5 %
|
|
Glycérol
|
.5 %
|
|
DTT
|
...1 mM
|
|
EDTA
|
100 mM
|
Une centrifugation est effectuée à 13000 g
à 4°C, pendant 20 min. Le surnageant obtenu contient les
protéines solubles. Il servira pour le dosage des protéines et
pour les tests des activités enzymatiques.
6.2. Dosage des protéines solubles
Le dosage des protéines est réalisé selon
la méthode de Bradford, (1976), utilisant le principe
de la liaison du bleu de Coomassie G250 avec les protéines. Ce
réactif coloré passe du violet au bleu lorsqu'il se lie à
la protéine et ainsi l'absorbance du complexe est lue. Ce complexe
colorant-protéine permet de déterminer la quantité en
protéines.
A un volume d'extrait protéique connu, 2 ml de bleu de
Coomassie sont ajoutés. Après 15 min, l'absorbance du
mélange est lue par spectrophotométrie à 595 nm. La
concentration protéique des échantillons est
déterminée à partir d'une gamme étalon de BSA
(Sigma) comprise entre 0 et 10ug.ml-1.
6.3. Détermination des activités des
enzymes antioxydantes
L'activité de la Catalase (CAT, EC 1.11.1.6) est
mesurée selon la méthode de Cakmak et Marschner,
(1992). Elle est testée par la mesure de la vitesse initiale de
disparition du peroxyde d'hydrogène, pendant 1 min à 240 nm, et
est calculée en utilisant le coefficient d'extinction de 39,4
mM-1 cm-1 pour le peroxyde d'hydrogène. Le tampon
réactionnel est composé de 50 mM de tampon phosphate (pH 7.0), 30
mM H2O2 et de l'extrait enzymatique.
La Gaïacol péroxydase (POD, GPX, EC 1.11.1.7) est
déterminée selon la méthode de Srinivas et
al., (1999), par suite de la formation de
tétra-gaïacol par la mesure de l'absorbance à 470 nm et en
utilisant le coefficient d'extinction de 26.6 mM-1 cm-1.
Le tampon réactionnel
Chapitre II Matériel et méthodes

18
(1 ml) contient 50 mM de tampon phosphate-gaïacol (pH
5,6), 30 mM H2O2 avec une aliquote de l'extrait de l'enzyme. La
réaction est effectuée pendant 2 min. Une unité de
péroxydase représente la quantité d'enzyme catalysant
l'oxydation de 1 ìmol de gaïacol.
L'activité de l'APX a été
déterminée selon la méthode de Cakmak et
Marschner, 1992, en mesurant la diminution de la densité de
l'ascorbate oxydé à 290 nm. Une unité de APX a
été définie comme la quantité d'enzyme
nécessaire pour consommer 1 ìmol ascorbate / min.
7. Extraction et dosage des composés
phénoliques
7.1. Préparation des extraits
L'extraction des composés phénoliques est
réalisée selon la méthode de Mau et al.,
(2001). Un gramme de matière sèche est
mélangé avec 10 mL méthanol. Le mélange est mis en
agitation pendant 60 min puis gardé au repos, pendant 24h à
4°C et à l'obscurité. Les extraits obtenus sont
filtrés avec des papiers filtres sans cendre puis conservés a
4°C pour servir par la suite aux dosages des polyphénols totaux,
des flavonoïdes et des activités antioxydante totale et
antiradicalaire.
7.2. Dosages des polyphénols totaux
Le dosage des polyphénols totaux est effectué
par un dosage colorimétrique selon la méthode de Folin Cicalteu
qui est un acide de couleur jaune constitué d'un mélange d'acide
phosphotungstique et d'acide phosphomolybdique (H3PMo12 O40). Ce mélange
se réduit lors de l'oxydation des polyphénols en un
mélange d'oxydes bleu de tungstène et de molybdène
(Ribereau-Gayon, 1968). L'intensité de cette coloration
bleue dont l'absorption maximale à 760nm renseigne sur la richesse de
l'extrait en polyphénols. Un volume de 125uL de l'extrait
méthanolique est mélangé avec 500 uL d'eau
distillée et 125 uL réactif de Folin Cicalteu. Après une
agitation vigoureuse du mélange suivie d'un repos de 3 min, on ajoute
1250uL d'une solution (Na)2CO3 à 7% et une prise de 1000uL d'eau
distillée est additionnée. Après un repos de 90 min
à l'obscurité et à la température ambiante; on
effectue une lecture de l'absorbance à 760 nm. Les teneurs des
polyphénols totaux sont déterminés à partir de la
courbe d'étalonnage linéaire réalisée par l'acide
gallique (0-200ug/mL), exprimées en milligrammes équivalents
d'acide gallique par gramme de matière sèche (mg EAG,
g-1MS) (Dewanto et al., 2002).
Chapitre II Matériel et méthodes

19
7.3. Dosage des flavonoïdes
a. Principe
Un dosage colorimétrique est basé sur la
formation d'un complexe, de couleur jaune, entre les flavonoïdes et le
trichlorure d'aluminium. La soude forme de son côté un autre
complexe qui rend la couleur rose dont l'intensité nous indique
l'importance du contenu de l'extrait en flavonoïdes (Zhishen et
al., 1999).
b. Mode opératoire
Un volume de 250 ul de l'extrait méthanolique est
mélangé avec 75 uL d'une solution de NaNO2 (5%). Après une
incubation à la température ambiante, on ajoute 150 uL d'une
solution de trichlorure d'aluminium à 10% (AlCl3,6H2O) fraîchement
préparé. Après cinq minutes de repos on ajoute 500 uL de
soude (NaOH, 1M) et on ajuste avec l'eau distillée jusqu'à 2.5 ml
(Dewanto et al., 2002). L'absorbance est
mesurée à 510 nm en se référant à un
témoin dépourvu de l'extrait. Les teneurs en flavonoïdes
sont calculées en utilisant une gamme étalon de catéchine
à des concentrations allant de 50 à 500 mg.l-1. Ces
teneurs en flavonoïdes sont exprimées en mg d'équivalent
catéchine par gramme de matière sèche (mg EC.
g-1MS).
7.4. Détermination de l'activité
antioxydante totale
a. Principe
Cette activité est mesurée par la
méthode qui consiste à réduire les ions Mo6+ en
Mo5+ par les extraits méthanoliques ainsi que la formation du
complexe (phosphate/ Mo5+) de couleur verte et à un pH acide
(Prieto et al., 1999).
b. Mode opératoire
Cette méthode consiste à ajouter 0.2 ml d'une
solution contenant de l'acide sulfurique (H2SO4; 0.6M), du phosphate de sodium
(NaH2PO4,H2O; 28mM) et de l'heptamolybdate d'ammonium ((NH4)6 MO7
O24, 4H2O; 4mM) à pH acide. Le mélange est ensuite
placé dans un bain - marie à 95°C, pendant 90 min.
Après refroidissement à la température ambiante,
l'absorbance est par la suite mesurée à 695 nm. L'activité
antioxydante totale est exprimée en mg d'équivalent acide
gallique par gramme de matière sèche (mg
EAG.g-1MS).
Chapitre II Matériel et méthodes

20
7.5. Pouvoir antiradicalaire (test DPPH)
a. Principe
C'est une méthode colorimétrique rapide, simple
et sensible, utilisée pour estimer l'activité antiradicalaire des
extraits. A une température ambiante, le radical synthétique le
2,2'-diphenyl-1-picrylhydrazyl (DPPHÿ) qui
présente en solution une coloration violette, disparaît au contact
d'une substance donneuse de protons.
DPPHÿ+Antioxydant-OH ? DPPH-H +
Aÿ
(Violette) (Incolore)
La cinétique de la dégradation de cette couleur
est détectable par le spectrophotomètre -UV à 517 nm par
rapport à un témoin. L'absorbance diminue en fonction de la
réduction du radical par les molécules antioxydantes
présentes dans l'extrait (Tadolini et al.,
2000).
b. Mode opératoire
Une solution mère de DPPH a été
préparée en mélangeant 15 mg DPPH dans 200 mL
méthanol pendant 2h a l'obscurité. Une prise d'essai de 0,75 mL
d'extrait dilué, du standard (0-0,1 mM ascorbate), ou blanc
(méthanol) est mise en présence de 1,5 mL de la solution de DPPH
à l'obscurité et pendant 30 min d'incubation. L'absorbance est
mesurée à 517 nm à l'aide d'un spectrophotomètre
UV-visible. Pour chaque échantillon, un blanc (méthanol) est
inclus a la place de DPPH.
8. Extraction et dosage des sucres solubles
totaux
La matière fraîche est extraite dans
l'éthanol 80% (v/v) à l'ébullition. Les sucres solubles
totaux sont dosés selon la méthode à l'anthrone
(Yemm et Willis, 1954) après incubation de l'extrait en
présence du réactif d'anthrone à 100°C pendant 10
minutes. La DO est mesurée à 640 nm contre une gamme croissante
de glucose.
9. Extraction et dosage de la proline
La proline est dosée selon la méthode de
Bates et al., (1973), mais dans la matière
végétale préalablement desséchée à
60°C. Elle est extraite par l'acide sulfosalicylique aqueux à 3%.
Le surnageant est récupéré après une centrifugation
effectuée à 14000 g, pendant 20 minutes à 4°C. La
proline est ensuite dosée dans le surnageant par une méthode
colorimétrique, qui est basée sur le complexe proline-ninhydrine,
obtenu après une heure à
Chapitre II Matériel et méthodes

21
Chapitre II Matériel et méthodes

22
100°C. Ce complexe coloré est ensuite extrait avec
du toluène et dosé à 520 nm par rapport à une gamme
étalon préalablement établie.
10. Extraction des lipides
a. Principe
Les lipides sont solubles à chaud ou à froid
dans les solvants organiques tels que l'éther de pétrole,
l'éther-di éthylique, l'hexane, l'acétone,
l'éthanol, le chloroforme, le méthanol, etc. En pratique l'hexane
et l'éther de pétrole à chaud sont les plus couramment
utilisés .On utilise pour cela un solvant à reflux dans un
extracteur de type SOXHLET. Les vapeurs chaudes du solvant traversent la
mouture (échantillon broyé) dans une cartouche, se condensent
plus haut dans un réfrigérant et retombent dans la cartouche
contenant la mouture. Lorsque le solvant remplit le corps de l'extracteur, il y
a siphonage et le solvant (chargé en huile fixe) retombe dans le ballon
d'ébullition. Le cycle continu jusqu'à l'extraction
complète de la matière grasse.
b. Mode opératoire
La détermination des matières grasses est faite
selon la méthode d'extraction par le SOXHLET, en utilisant l'hexane
comme solvant. On pèse 5g de feuilles du ricin bien broyées dans
une cartouche, on le recouvre avec coton et on la place dans le Soxhlet. Peser
le ballon qui servira à recouvrir le solvant et y introduire 50 ml
hexane.

Schéma de SOXHLET
Ensuite on va chasser par distillation la majeure partie du
solvant à l'aide de l'évaporateur rotatif (ROTAVAPOR) pour
éviter l'ébullition de l'huile qui à la longue pourrait
modifier les indices d'acidité.

Schéma de ROTAVAPOR
10.1. Détermination du rendement en matière
grasse
La teneur en huile ou teneur en matière grasse totale,
exprimée en pourcentage de la matière sèche, est
calculée selon la formule suivante :
Teneur en matières grasses (%) = ((P1 - P0)*100) /
PS
Avec :
P0 : poids en g du ballon à vide
P1 : poids en g du ballon contenant les lipides
PS : poids en g de la matière sèche
10.2. Détermination de la teneur en chlorophylle
et en béta carotène
On pèse 0,6g de l'échantillon dans une fiole de
25 ml et on complète avec le cyclohexane jusqu'à le trait de
jauge, puis on fait une agitation jusqu'à l'obtention d'une solution
limpide. Ensuite une lecture de DO à 670 nm pour la chlorophylle et
l'autre 470 pour le /3-carotène.
a. Teneur en chlorophylle
Teneur en chl:
absorbance670*106/E1*100*d
Avec :
E1 = 613 (l'extinction spécifique)
d = 1 cm (l'épaisseur de cuve)
b. Teneur en /3-carotène
Teneur en /3-carotène:
absorbance470*106/E2*100*d
Chapitre II Matériel et méthodes

23
Avec :
E2 = 2000 (l'extinction spécifique)
d = 1 cm (l'épaisseur de cuve)
11. Analyse chromatographique des acides gras
Les acides gras (AG) pour être analysés par
chromatographie en phase gazeuse doivent être transformés
chimiquement en des composés volatils, les esters méthyliques
d'acides gras (EMAGs). Cette réaction chimique s'appelle la
méthylation. La méthode utilisée est une
trans-méthylation alcaline directe (ISO 12966-2 : 2012),
illustrée par l'équation suivante :
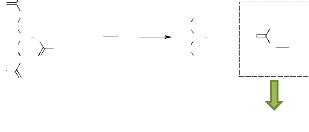
H2C
R
O
O
R
H2C
CH
O
O
OH
R
Glycérol
O
Triglycéride
OH
H2C
EMAGs
CH3
OH-
+ 3
O
Méthanol
CH
R
O
GC
O
La séparation des différents esters
méthyliques d'acides gras (EMAG) est effectuée par
chromatographie en phase gazeuse en utilisant un appareil de marque HP-6980
(Agilent Technologies, Palo Alto, CA, USA) série II muni d'un
détecteur à ionisation de flamme (FID), d'un auto-injecteur et
d'un échantillonneur automatique.
R: Chaîne carbonée de l'acide gras
La séparation des différents EMAG a
été réalisée sur une colonne capillaire polaire
TR-FAME (60 m de longueur, 0,32 mm de diamètre, 0,25 um
d'épaisseur du film). La température du four a été
programmée de la manière suivante: isotherme à 170°C
(2 min), augmentation de 3°C/min jusqu' à 240°C, de 170°C
à 240°C à raison de 3°C/mn pendant 15 mn, isotherme
à 240°C pendant 10mn. La température de l'injecteur et du
détecteur est maintenue à 225°C.
Les analyses ont été menées en mode split
(Rapport de split : 60/l, gaz vecteur: hélium, Débit total: 1.6
ml/min). Le système est piloté par un logiciel type Chemstation
qui assure l'intégration électronique des différents
pics.
Chapitre II Matériel et méthodes

24
L'identification des EMAG a été effectuée
en comparant leurs temps de rétention avec ceux des étalons de
référence acheté chez Fluka. Les compositions des EMAG (%)
se réfèrent au rapport de pourcentage de chaque composant au
total des acides gras. Le "double bond index" (DBI) a été
calculé comme suit (Gignon et al, 2004):
Avec, AM : acides monoénoïques ;
AD : acides diénoïques et AT :
acides triénoïques
ODR: Oleic desaturase ratio ([(C18:2+C18:3) /
(C18:1+C18:2+C18:3)]X100);
LDR: Linoleic desaturase ratio
([C18:3 / (C18:2+C18:3)] X100)
12. Analyse statistique
Toutes les expériences ont été
réalisées trois fois au cours de trois expériences
successives. Les résultats ont été exprimés par une
moyenne plus ou moins l'écart type.
| 

